
Research Article | DOI: https://doi.org/10.31579/2690-4861/542
College of Chemistry and Material Science, Shandong Agricultural University, Taian 271018, P. R. China.
*Corresponding Author: Nan Lu, College of Chemistry and Material Science, Shandong Agricultural University, Taian 271018, P. R. China.
Citation: Nan Lu and Chengxia Miao, (2024), Theoretical investigation on Friedel−Crafts reaction followed by rear-rangement/aromatization or ring-opening delivering benzoheterocycle and polycyclic alcohol, biaryl carboxylic acid, International Journal of Clinical Case Reports and Reviews, 19(2); DOI:10.31579/2690-4861/542
Copyright: © 2024, Nan Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 16 September 2024 | Accepted: 30 September 2024 | Published: 10 October 2024
Keywords: biaryl carboxylic acid; friedel−crafts; aromatization; ring-opening; benzoheterocycle
Our DFT calculations provide the first theoretical investigation on CF3SO3H-promoted [1,5]Friedel−Crafts of 2-aryoxy-1,3-indandione and base-facilitated [1,6]Friedel−Crafts of 1,3-dicarbonyl. An intramolecular [1,5]Friedel−Crafts addition took place by activating carbonyl through H bridge with CF3SO3H. The resulting tertiary alcohol underwent dehydration producing reactive carbocation, which instigated a cascade of carbox-yl group formation, ring-opening via C−C bond dissociation and C=C bond formation realizing aromatization. The product 3-aryl-2-benzo was yielded with recovered CF3SO3H. The electron-rich phenoxy group was deprotonated by HO- forming water. The initial nucleophilic addition to spatial-adjacent carbonyl afforded dieneone. It isomerized to the first product polycyclic alcohol. Then carboxylation occurred via hydroxyl shift followed by ring-opening aromatization to the second product biaryl carboxylic acid. The positive solvation effect is suggested by decreased absolute and activation energies in solution compared with in gas. These results are supported by Multiwfn analysis on FMO composition of specific TSs, and MBO value of vital bonding, breaking.
As privileged structural components in biologically active compounds, the benzofuran and indole motifs are important in pharmaceutical science and drug industry due to versatile biological properties and chemical activities [1]. In this field, the benzofuran derivatives are especially significant as potential therapeutic agents. For instance, Shang researched the antioxidant activity of viniferifuran [2]. Song discovered aromatic polyketides from deep-sea cold-seep mussel associated endozoic fungus talaromyces minioluteus CS-138 [3]. Cao obtained cytotoxic alkaloids from micromelum integerrimum [4]. However, traditional approaches often require multistep or complex reaction condition involving cascade reaction with α-imino Gold carbenes and Pd-catalyzed asymmetric Larock indole synthesis [5,6]. There fore the intramolecular C−N construction techniques offer efficient and sustainable routes [7,8] such as Wang’s iron-catalyzed intramolecular C−H amination and He’s Palladium-catalyzed enantioselective cacchi reaction for synthesis of N−H carbazoles and axially chiral 2,3-disubstituted indoles [9,10].
On the other, biaryl carboxylic acids are also key structural motifs in natural products for synthesis of axially chiral compounds. Biaryl lactone was used for derivation of potential antimycotic agents against Candida strains [11]. In addition, binaphthyl scaffold like MeO-BINA-Cox is a class of versatile structure in asymmetric C−H functionalization [12]. Li prepared optically active 2,2’-Dibromo-6,6’-diiodo-1,1’-biphenyl as powerful precursor for modular synthesis of functionalized atropisomers [13]. Wei realized Fe-catalyzed difunctionalization of aryl titanates enabled by Fe/Ti synergism [14]. Linde developed atroposelective brominations to access chiral biaryl scaffolds applying high-valent Pd-catalysis [15]. Ansari utilized trichloromethyl carbanion in aqueous micelles to access carboxylic Acids from (Hetero)aryl halides [16]. Many efforts have been dedicated to these structures such as Palladium/norbornene-catalyzed decarbonylative difunctionalization of thioesters [17]. Despite with achievements of photoinduced protocol for aerobic oxidation of aldehydes to carboxylic acids, claisen approach to 4’-Ed4T and Palladium-catalyzed C-H ortho arylation of benzoic acids with diaryliodonium salt [18-20].Some limitations still hampered owing to the requirement of prefunctionalization.
Tong found phosphine-catalyzed (4 + 2) annulations of δ-acetoxy allenoates and ketones to construct 1,3-cyclohexadienes [21]. Aspired by this, Liu developed [1,6]-type Friedel−Crafts reaction resulting in polycyclic alcohol then transformed into biaryl carboxylic acids through ring-opening rearrangement [22]. The 1,3-dicarbonyls were used to accomplish selective C−C bond formation previously [23,24]. One of the carbonyl groups is integrated into product structure. A recent progress was constrained [1,5]-type Friedel−Crafts reaction/rearrangement/aromatization process to synthesize 2-substituted-3-aryl benzofuran from easily available 2-aryoxy-1,3-indandione [25]. Although a range of benzoheterocycles were yielded, many problems still puzzled and there was no report about detailed mechanistic study explaining the promotion of CF3SO3H as Bronsted acid (BA). How the reactive carbocation instigated cascade C−C bond dissociation and C=C bond formation? Why base was necessary for nucleophilic addition of electron-rich phenoxy group to spatial-adjacent carbonyl in generation of dieneone? What’s specific process of ring-opening rearrangement and aromatization? To solve these questions in experiment, an in-depth theoretical study was necessary for this strategy also focusing on the comparison of [1,6]-, [1,5]-type Friedel−Crafts and excellent regio-, diastereoselectivity.
Optimized structures were obtained at M06-2X/6-31G(d) level of theory with GAUSSIAN09 [26]. In tests of popular DFT methods [27], M06-2X functional attained smaller standard deviation of difference between calculated value and experimental value in geometries than B3LYP including Becke's three-parameter hybrid functional combined with Lee−Yang−Parr correction for correlation [28,29]. The best compromise between accuracy and time consumption was provided with 6-31G(d) basis set on energy calculations. Also, M06-2X functional was found to give relatively accurate results for catalysed enantioselective (4 + 3), concerted [4 + 2], stepwise (2 + 2) cycloaddition and catalysed Diels−Alder reactions [30,31]. Together with the best performance on noncovalent interaction, M06-2X functional is believed to be suitable for this system [32-34]. The nature of each structure was verified by performing harmonic vibrational frequency calculations. Intrinsic reaction coordinate (IRC) calculations were examined to confirm the right connections among key transition-states and corresponding reactants and products. Harmonic frequency calculations were carried out at the M06-2X/6-31G(d) level to gain zero-point vibrational energy (ZPVE) and thermodynamic corrections at 353 K and 1 atm for each structure in acetonitrile.
The solvation-corrected free energies were obtained at the M06-2X/6-311++G(d,p) level by using integral equation formalism polarizable continuum model (IEFPCM) in Truhlar’s “density” solvation model [35-39] on the M06-2X/6-31G(d)-optimized geometries. As an efficient method obtaining bond and lone pair of a molecule from modern ab initio wave functions, NBO procedure was performed with Natural bond orbital (NBO3.1) to characterize electronic properties and bonding orbital interactions [40-42]. The wave function analysis was provided using Multiwfn_3.7_dev package [43] including research on frontier molecular orbital (FMO) and Mayer bond order (MBO).
The mechanism was explored for (a) CF3SO3H-promoted [1,5] Friedel−Crafts of 2-aryoxy-1,3-indandione 1 leading to 2-substituted-3-aryl benzofuran 2; (b) Base-facilitated [1,6]Friedel−Crafts of 1,3-dicarbonyl I resulting in polycyclic alcohol 3 then transformed into biaryl carboxylic acid 4 (Scheme 1). Illustrated by black arrow of Scheme 2a, an intramolecular [1,5]Friedel−Crafts addition of 1,3-indandione 1 took place by activating carbonyl through H bridge with CF3SO3H as Bronsted acid. Then the resulting tertiary alcohol in3 underwent dehydration assisted by CF3SO3H to produce reactive carbocation in4, which then instigated a cascade of carboxyl group formation, ring-opening via C−C bond dissociation and C=C bond formation realizing aromatization. The final product 3-aryl-2-benzo 2 was yielded binding recovered CF3SO3H. Shown by black arrow of Scheme 2b, the electron-rich phenoxy group was deprotonated under the catalysis of base HO- forming water.The initial nucleophilic addition underwent with spatial-adjacent carbonyl resulting in dieneone II after removal of HO-. Subsequently, II isomerized to the first product polycyclic alcohol 3, from which carboxylation proceeds via hydroxyl shift rearrangement followed by ring-opening aromatization leading to the second product biaryl carboxylic acid 4. The schematic structures of optimized TSs in Scheme 2 were listed by Figure 1. The activation energy was shown in Table 1 for all steps. Supplementary Table S1, Table S2 provided the relative energies of all stationary points. According to experiment, the Gibbs free energies in acetonitrile solution phase are discussed here.
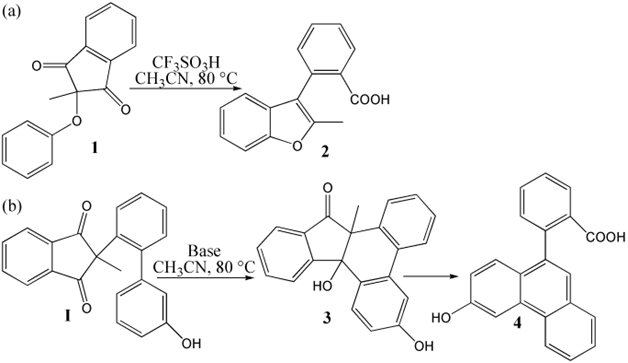
Scheme 1 (a) CF3SO3H-promoted [1,5] Friedel−Crafts of 2-aryoxy-1,3-indandione 1 leading to 2-substituted-3-aryl benzofuran 2; (b) Base-facilitated [1,6] Friedel−Crafts of 1,3-dicarbonyl I resulting in polycyclic alcohol 3 then transformed into biaryl carboxylic acid 4.
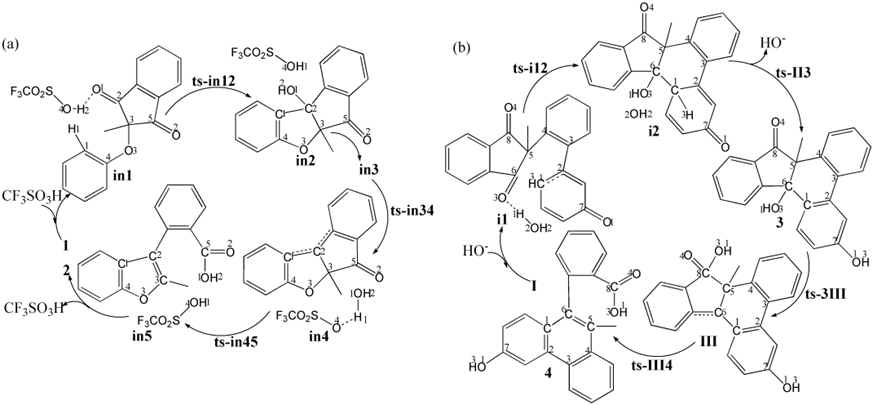
Scheme 2: Proposed reaction mechanism of TS is named according to the two intermediates it connects.
| Species | ΔGgas | ΔGsol(CH3CN) |
| 1 | 0.00 | 0.00 |
| 2 | -25.36 | -29.57 |
| 1+CF3SO3H | 0.00 | 0.00 |
| in1 | -16.53 | -4.90 |
| ts-in12 | 13.57 | 11.25 |
| in2 | -28.20 | -16.00 |
| in3 | -1.71 | 7.71 |
| ts-in34 | 19.91 | 14.22 |
| in4 | 6.93 | -0.52 |
| ts-in45 | 13.12 | 0.54 |
| in5 | -39.98 | -31.98 |
| Species | ΔGgas | ΔGsol(CH3CN) |
| I+OH | 0.00 | 0.00 |
| i1 | -58.08 | -10.32 |
| ts-i12 | -48.44 | -1.15 |
| i2 | -62.84 | -14.39 |
| I | 0.00 | 0.00 |
| II | 11.20 | 1.36 |
| ts-II3 | 44.52 | 30.43 |
| 3 | -2.96 | -9.64 |
| ts-3III | 22.10 | 11.65 |
| III | 8.95 | 3.38 |
| ts-III4 | 21.23 | 13.19 |
| 4 | -20.81 | -26.93 |
Table S1. Calculated relative energies (all in kcal mol-1, relative to isolated species) for the ZPE-corrected Gibbs free energies (ΔGgas), Gibbs free energies for all species in solution phase (ΔGsol) at 353 K by M06-2X/6-311++G(d,p)//M06-2X/6-31G(d) method and difference between absolute energy.
| TS | ΔG≠gas | ΔG≠sol |
| ts-in12 (360i) | 30.1 | 16.1 |
| ts-in34 (292i) | 21.6 | 6.5 |
| ts-in45 (243i) | 6.2 | 1.1 |
| ts-i12 (248i) | 9.6 | 9.2 |
| ts-II3 (1812i) | 33.3 | 29.1 |
| ts-3III (377i) | 25.1 | 21.3 |
| ts-III4 (100i) | 12.3 | 9.8 |
Table S2. The activation energy (local barrier) (in kcal mol−1) of all reactions in the gas, solution phase calculated with M06-2X/6-311++G(d,p)//M06-2X/6-31G(d) method.
| TS | ΔG≠gas | ΔG≠sol |
| ts-in12 | 30.1 | 16.2 |
| ts-in34 | 21.6 | 6.5 |
| ts-in45 | 6.2 | 1.1 |
| ts-i12 | 9.6 | 9.2 |
| ts-II3 | 33.3 | 29.1 |
| ts-3III | 25.1 | 21.3 |
| ts-III4 | 12.3 | 9.8 |
Table 1 The activation energy (in kcal mol−1) of all reactions in gas and solvent
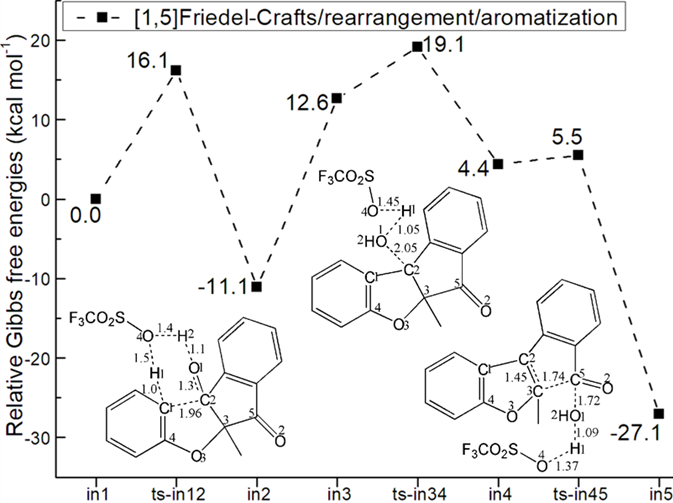
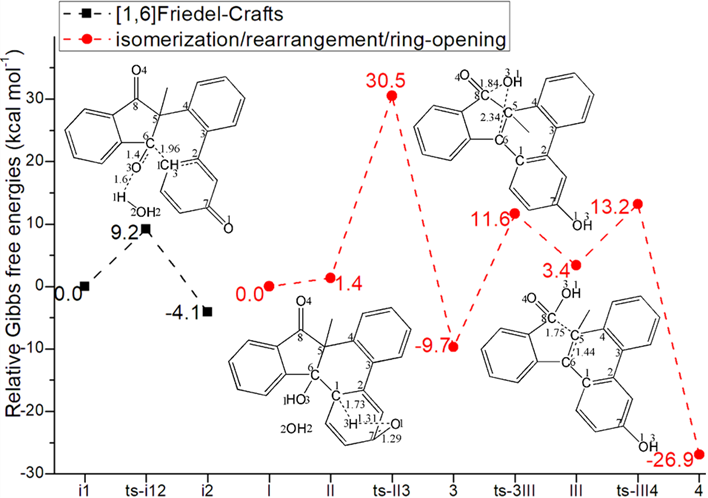
Figure 1: Relative Gibbs free energy profile in solvent phase starting from complex (a) in1 (b) i1, I (Bond lengths of optimized TSs in Å).
3.1 [1,5] Friedel-Crafts/rearrangement/aromatization
The initial complex is located as in1 involving H bond stabilization between CF3SO3H and carbonyl of 1. An intramolecular [1,5]Friedel−Crafts addition took place via ts-in12 in step 1 with the activation energy of 16.1 kcal mol−1 relative to starting point in1 exothermic by -11.1 kcal mol−1 producing tertiary alcohol in2 (black dash line of Figure 1a). The transition vector includesdual proton transfer C1···H1···O4, O4···H2···O1, resultant stretching of C2-O1 double bond to single one and the slightly delayed nucleophilic addition of C1 to C2 (1.0, 1.5, 1.4, 1. 1, 1.3, 1.96 Å) (Figure S1a). Obviously, the driving force is attributed by H bridge from CF3SO3H via simultaneously activating nucleophilic C1 and carbonyl C2=O1. Once C1-C2 is bonded, carbonyl becomes hydroxyl O1H2 in the new five membered ring of in2, which transformed to in3 more reactive with relative energy increased by 23.7 kcal mol−1 ready to initiate next step.
Then assisted by CF3SO3H, the resulting tertiary alcohol in3 undergoes dehydration via ts-in34 as step 2 with activation energy of 6.5 kcal mol−1 endothermic by 4.4 kcal mol−1 producing reactive carbocation in4. The transition vector suggests that the obtained proton H1 from C1 in previous step 1 is handed over to hydroxyl O1H2 by O4 of CF3SO3H to assemble water molecule H1-O1H2. This causes the breaking of C2-O1 single bond concertedly (1.45, 1.05, 2.05 Å) (Figure S1b). The leaving of O1H2 from C2 makes it positive and sp2 hybrid in carbocation in4, where there is still O4···H1O1 H bonding.
At last, a cascade rearrangement/aromatization takes place via ts-in45 in subsequent step 3 with low activation energy of 1.1 kcal mol−1 affording in5 exothermic by -27.1 kcal mol−1. The transition vector is complicated contains a series of atomic motion. On one hand, the water returns H1 to O4 of CF3SO3 faciliting its recovery via O1···H1···O4 (1.09, 1.37 Å). The hydroxyl O1H2 is thus approaching another carbonyl C5=O2 via O1···C5 (1.72 Å) forming carboxyl group. On the other, C3−C5 bond is dissociated inducing ring-opening and C2=C3 double bond formation realizing aromatization (1.74, 1.45 Å) (Figure S1c). The final in5 is rather stablecombining recovered CF3SO3H and product 3-aryl benzofuran involving carboxyl moiety. Ultimately, the [1,5]Friedel−Crafts in step1 is determined to be rate-limiting for CF3SO3H-promopted whole process.
3.2 [1,6] Friedel−Crafts/isomerization/carboxylation/ring-opening aromatization
As a comparison, [1,6]Friedel−Crafts was also explored from 1,3-dicarbonyl I facilitated by base HO- (black dash line of Figure 1b). In initial i1, the electron-rich phenoxy group was deprotonated by additional HO- forming water H1-O2H2, which activated spatial-adjacent carbonyl via H bonding. Therefore, the nucleophilic addition from C1 to C6 occurs readily via ts-i12 in step 1 with activation energy of 9.2 kcal mol−1 exothermic by -4.1 kcal mol−1 resulting in i2 delivering dieneone intermediate II after removal of HO-. The transition vector corresponds to the approaching of H1 to O3, elongation of carbonyl C6-O3 from double bond to single and C1-C6 linkage (1.6, 1.4, 1.96 Å) (Figure S1d). The typical C1-C6 bond gives new six membered ring in stable i2.
Without HO-, II turns to be more reactive with higher relative energy (1.4 kcal mol−1) than I. Subsequently, the isomerizationof II in step 2 happens via ts-II3 with activation energy of 29.1 kcal mol−1 exothermic by -9.7 kcal mol−1 generating 3 (red dash line of Figure 1b). The transition vectorreveals detailed atomic motion comprising proton H3 transfer from C1 to O1 and the resultant stretching of carbonyl C7-O1 from double to single (1.73, 1.31, 1.29 Å) (Figure S1e). As the first product polycyclic alcohol, 3 involves the recovered phenolic hydroxyl group O1H3.
In next step 3, the rearrangement via hydroxyl shift proceeds via ts-3III with activation energy of 21.3 kcal mol−1 yielding cation III stabilized by two conjugated phenyl ring endothermic by 3.4 kcal mol−1. This process is illustrated according to the transition vector composed of hydroxyl O3H1 leaving from C6 to C8 that is C6···O3 breaking and C8···O3 bonding (2.34, 1.84 Å) (Figure S1f). The outcome not only includes formation of reactive cation at C6 but carboxylation producing new carboxyl group O4=C8-O3H1.
From reactive III, the final ring-opening aromatization is easy to be initiate via ts-III4 in step 4 with a low barrier of only 9.8 kcal mol−1 exothermic by -26.9 kcal mol−1 affording4 as the second product biaryl carboxylic acid. This step is quite favorable both from kinetics and thermodynamics. Demonstrated by the transition vector, the ring-opening is accomplished via C5···C8 cleavage and aromatization through C5-C6 shortened from single bond to double (1.75, 1.44 Å) (Figure S1g). Comparatively, the isomerizationof dieneone in step 2 is determined to be rate-limiting for base-facilitated [1,6]Friedel−Crafts case.
To highlight the idea of feasibility for changes in electron density and not molecular orbital interactions are responsible of the reactivity of organic molecules, quantum chemical tool Multiwfn was applied to analyze of electron density such as MBO results of bonding atoms and contribution of atomic orbital to HOMO of typical TSs (Table S3, Figure S2). These results all confirm the above analysis.
| O4···H2 | H2···O1 | C1···C2 | C2···O1 | ||
ts-in12 | 0.23 | 0.45 | 0.45 | 1.29 | |
| O4···H1 | H1···O1 | C2···O1 | |||
| ts-in34 | 0.22 | 0.48 | 0.28 | ||
| O1···H1 | H1···O4 | O1···C5 | C5···C3 | C3···C2 | |
| ts-in45 | 0.43 | 0.25 | 0.45 | 0.64 | 1.27 |
| H1···O3 | C6···O3 | C1···C6 | |||
| ts-i12 | 0.17 | 1.37 | 0.50 | ||
| C1···H3 | H3···O1 | C7···O1 | |||
| ts-II3 | 0.39 | 0.39 | 1.34 | ||
| C6···O3 | C8···O3 | ||||
| ts-3III | 0.23 | 0.43 | |||
| C5···C8 | C5···C6 | ||||
| ts-III4 | 1.24 | 0.51 |
Table S3. Mayer bond order (MBO) of typical TSs


Figure S1. Evolution of bond lengths along the IRC for (a) ts-in12 (b) ts-in34 (c) ts-in45 (d) ts-i12 (e) ts-II3 (f) ts-3III (g) ts-III4 at M06-2X/6-311++G(d,p) level.
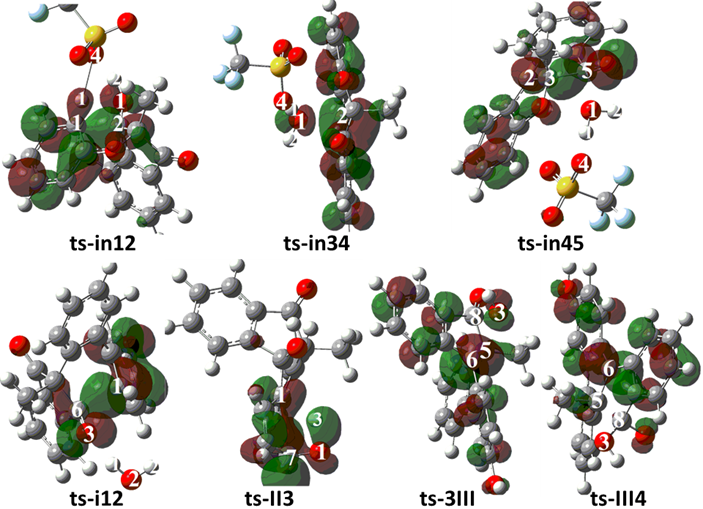
Figure S2. Highest Occupied Molecular Orbital (HOMO) of typical TSs. Different colors are used to identify the phase of the wave functions.
Our DFT calculations provide the first theoretical investigation on CF3SO3H-promoted [1,5]Friedel−Crafts of 2-aryoxy-1,3-indandione leading to 2-substituted-3-aryl benzofuran and base-facilitated [1,6]Friedel−Crafts of 1,3-dicarbonyl resulting in polycyclic alcohol then transformed into biaryl carboxylic acid. For the former, an intramolecular [1,5]Friedel−Crafts addition of 1,3-indandione took place by activating carbonyl through H bridge with CF3SO3H. The resulting tertiary alcohol underwent dehydration assisted by CF3SO3H producing reactive carbocation, which then instigated a cascade of carboxyl group formation, ring-opening via C−C bond dissociation and C=C bond formation realizing aromatization. The final product 3-aryl-2-benzo was yielded binding recoveredCF3SO3H. For the latter, the electron-rich phenoxy group was deprotonated under the catalysis of HO- forming water. The initial nucleophilic addition underwent with spatial-adjacent carbonyl affording dieneone after removal of HO-. Subsequently, dieneone isomerized to the first product polycyclic alcohol, from which carboxylation proceeds via hydroxyl shift rearrangement followed by ring-opening aromatization leading to the second product biaryl carboxylic acid. Comparatively, [1,5]Friedel−Crafts in step 1 is determined to be rate-limiting for CF3SO3H-promopted process. While the isomerization of dieneone in step 2 is rate-limiting for base-facilitated [1,6]Friedel−Crafts case. The positive solvation effect is suggested by decreased absolute and activation energies in acetonitrile solution compared with in gas. These results are supported by Multiwfn analysis on FMO composition of specific TSs, and MBO value of vital bonding, breaking.
Supplementary data available: [Computation information and cartesian coordinates of stationary points; Calculated relative energies for the ZPE-corrected Gibbs free energies (ΔGgas), and Gibbs free energies (ΔGsol) for all species in solution phase at 353 K.]
Conceptualization, Nan Lu; Methodology, Nan Lu; Software, Nan Lu; Validation, Nan Lu; Formal Analysis, Nan Lu; Investigation, Nan Lu; Resources, Nan Lu; Data Curation, Nan Lu; Writing-Original Draft Preparation, Nan Lu; Writing-Review & Editing, Nan Lu; Visualization, Nan Lu; Supervision, Chengxia Miao; Project Administration, Chengxia Miao; Funding Acquisition, Chengxia Miao. All authors have read and agreed to the published version of the manuscript.
This work was supported by National Natural Science Foundation of China (21972079) and Key Laboratory of Agricultural Film Application of Ministry of Agriculture and Rural Affairs, P.R. China.
The authors declare no conflict of interest.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
