
Short Communication | DOI: https://doi.org/10.31579/2637-8914/136
Biochemical Technology Program, Dhamar University, Yemen.
*Corresponding Author: Haitham Ahmed Al-Madhagi, Biochemical Technology Program, Dhamar University, Yemen.
Citation: Haitham Ahmed Al-Madhagi (2023), Metabolic memory of type 1 diabetes revisited, J. Nutrition and Food Processing, 6(4); DOI:10.31579/2637-8914/136
Copyright: © 2023, Haitham Ahmed Al-Madhagi. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 03 April 2023 | Accepted: 01 June 2023 | Published: 15 June 2023
Keywords: .
.
In 2021, there were 8.4 million cases of type 1 diabetes mellitus (T1DM) worldwide which projected to reach 17.4 million cases in 2040 [1]. Patients diagnosed with T1DM are on life-long insulin injections in order to regulate their blood glucose levels. The standard insulin therapy and subsequent monitoring HbA1c is no longer sufficient for T1DM patients management. Instead, postprandial hyperglycemia as well as glycemic variations should be minimized to reach near normoglycemic state. To do so, intensive insulin therapy should be introduced so as to restore homeostatic glycemic control [2]. Unfortunately, keeping HbA1c in the allowed range is alas not adequate to reduce the incidence of macro and microvascular complications of T1DM due the emerging "metabolic memory" [3].
The "metabolic memory" terminology was coined for the first time in 1987 documenting the relatively faster development of T1DM complications despite the control of blood glucose and HbA1c levels. This was followed by successive clinical trials demonstrating the reduced risk of suffering from complications in intensively-controlled patients as compared to regular therapy confirming the existence of such metabolic memory. Metabolic memory can be defined as the highly adaptable behavior of body to the external intervention in the first period after diabetes has established. There are 4 mechanisms that are most likely to be responsible for the formation of metabolic memory: oxidative stress, advanced glycated end products (AGE), epigenetic modifications, and chronic inflammation (Fig 1) [4]. Although oxidative stress and its consequence, AGEs, are contributing factors, interventions intended to block AGE formation failed to 'erase' metabolic memory consequences. Reactive species generation, the driving fuel of oxidative stress, is unavoidable within human body. In addition, hyperglycemia-induced reactive oxygen species generation also leads to activation of nuclear factor kappa B (NF-κB) which in turn triggers protein kinase C exaggerating the oxidative events. In result, NO and lipid peroxides are formed in smooth muscle cells, endothelial cells and adipocytes causing thereby low-grade inflammatory response [5]. In fact, oxidative stress and inflammation are firmly associated, i.e. oxidative stress signals the activation of different transcriptional factors that ultimately results in the differential expression of inflammation-inducing genes and, in contrast, inflammation feeds the oxidative stress vicious cycle [6].
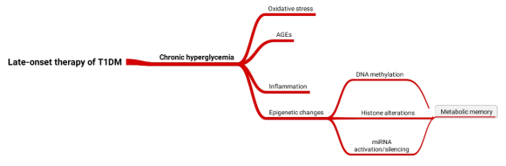
Figure 1: Mechanisms underlie the formation of metabolic memory.
Indeed, it has been proven that what lies behind the several, if not all, forms of long-term memory and adaptation to an effector is the epigenetic changes. These include 3 main players: DNA methylations, histone covalent modifications and miRNA biogenesis and action. Maternal nutritional status during pregnancy deeply influences fetal predisposition to T1DM. Additionally, fetal "critical period" which is the first 7 years after born, determines to a great extent the likelihood of disease development. Histone covalent modifications takes the largest part of implication to metabolic memory as represented by the many histone-modifying enzymes. Such changes take place in interplaying cells involving smooth muscle cells, endothelial cells, retinal and renal cells [3]. The good news here is that epigenetic alterations are reprogrammable, i.e. can be targeted by certain inhibitors so as to erase the formed memory. Hypermethylation of CpG islands in DNA of patients with end-stage kidney failure and albuminuria was found. Similarly, upon putting aortic endothelial cells in hyperglycemic stress, H3K4me1 mark was persistently detected in the promoter of NF-κB even though the cells had been removed from hyperglycemic state [7]. Moreover, monocytes isolated from patients with a history of elevated HbA1c levels had more promotor regions enriched with H3K9Ac alterations. These promoters belong to genes related to numerous diabetes and diabetic complication-implicated pathways, including the TNFR2 signaling as well as the NF-κB pathway. Likewise, plenty of miRNAs were shown to contribute to the foundation of metabolic memory. miR-21, miR-192, miR-214, and miR-377 were observed in the kidneys of T1DM patients. miR-21 triggered fibronectin expression and renal cell hypertrophy through the activation of TORC1 activity. In addition, reduced PTEN levels were also correlated with increased expression of miR-214 as a consequence of hyperglycemia [7].
In conclusion, this highlights the pivotal role of the components of epigenetic alterations that underlie the development of metabolic memory in T1DM patients. Furthermore, targeting these components should be expanded to reach conclusive result regarding the late clinical intervention of T1DM which creates a hope to those patients.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
