
Case Report | DOI: https://doi.org/10.31579/2692-9392/085
1 Pediatric Hepato-Gastroenterology and Nutrition Department, Rabat Children's Hospital, Faculty of Medicine and Pharmacy, Mohammed V Rabat University, Morocco
2 Anatomopathology Department of the Rabat Children's Hospital, Rabat Children's Hospital, Faculty of Medicine and Pharmacy, Mohammed V University Rabat, Morocco
*Corresponding Author: Khadija Mouaddine, Pediatric Hepato-Gastroenterology and Nutrition Department, Rabat Children's Hospital, Faculty of Medicine and Pharmacy, Mohammed V Rabat University, Morocco.
Citation: Khadija Mouaddine, Mouna Sabib, Laila Benbella, Najat Lamalmi, Nezha Mouane (2022). Waldmann’s Disease Revealed by Chronic Diarrhea and Lymphedema: Case Report. J. Archives of Medical Case Reports and Case Study, 5(2); DOI:10.31579/2692-9392/085
Copyright: © 2022 Khadija Mouaddine, This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 25 November 2021 | Accepted: 27 December 2021 | Published: 07 January 2022
Keywords: intestinal lymphangiesctasia; waldmann disease; lymphedema
Intestinal lymphangiesctasia (IL) is a rare disease characterized by the dilation of intestinal lymphatics. It can be classified as primary or secondary depending on the underlying etiology. Usually seen in childhood or adolescence, chronic diarrhea and diffuse edema are the main clinical manifestations of the disease. True lymphoedemas can also be present and affect the lower and upper limbs, they are concomitant with the diagnosis or occur during the course.
The diagnosis is based on the visualization of duodenal lymphangiesctasia. The primitive nature of the disease being affirmed by the elimination of the diseases causing secondary lymphangiectasias. Treatment is based on a strict hypo lipid diet enriched with medium chain triglycerides (MCTs).
We present through this work the observation of a 12-month-old infant who presented with primary intestinal lymphangiesctasia revealed by chronic diarrhea with lymphoedema of the left upper limb.
Intestinal lymphangiesctasia (IL) is a rare and benign disease characterized by focal or diffuse dilation of mucous, sub mucosal and subserosal lymphatics [1]. In addition to being an important cause of protein loss [2], it is frequently associated with extra intestinal lymphatic abnormalities [3].
Depending on the underlying pathology, lymphangiectasia can be classified as primary or secondary disease. The primary form (PIL) probably represents a congenital disorder of the mesenteric lymphatics, as for the secondary form, it may be secondary to diseases such as constrictive pericarditis, lymphoma, sarcoidosis and scleroderma [4].The modification of the diet is the mainstay of the management of this pathology with a variable response.
The objective of our work is to describe the observation of a female infant who presented with primary intestinal lymphangiesctasia revealed by chronic diarrhea with lymphedema of the left upper limb.
Patient and Observation
Our patient is a 12-month-old female infant, from an out-of-consanguineous marriage with healthy siblings, and a well-followed pregnancy. The delivery was vaginally with birth weight = 3400g, exclusive breastfeeding for 02 months then mixed breastfeeding, diversified at the age of 07 months. She has had diarrhea since the age of 6 months at a rate of 6 stools / day. The stools are watery, there is no mucus or blood, and there is progressive abdominal distension and lymphedema of the left upper limb. There are no extra digestive signs, including no respiratory signs and no recurrent infections.
On physical examination, we find a haemodynamically and respiratory stable infant, pale, with a weight of 10kg (M) and a height of 75cm (M). There is no dysmorphia. The abdomen is distended without hepatomegaly or splenomegaly, with the presence of lymphedema of the left hand (Figure 1). No jaundice, no edema, the urine dipstick is negative.

On the biological level: there is a hypoproteinemia at 27g / l with hypo albuminemia at 15g / l. Calcemia is 72mg / l, ferritin is reduced to 8 ng / ml, total cholesterol at 1.37g / l, HDL at 0.27g / l LDL at 0.96 g / l, triglycerides at 0.72 g / l. The liver function tests are normal with ASAT at 47 U / L, ALAT at 18 U / L, GGT at 4 U / L and PAL = 173 .TP at 100%. The blood count reveals a hemoglobin level of 11.1 g / dl with VGM at 73.7 µm3 and CCMH at 31.7%, a leukocyte level at 7060 / µl with polymorphonuclear neutrophils at 4780 / µl and lymphocytes at 1140 / µl and eosinophils at 210 / µl with thrombocytosis at 626000 / µl.
The blood ionogram as well as the renal function are normal, the anti-transglutaminase antibodies are normal with a level of Ig A = 1.25 AU / ml and Ig G = 0.41 AU / ml.
Radiologically: the echo Doppler of the left upper limb as well as the echoceur are normal, the abdominal ultrasound showed the presence of a thickened and agglutinated aspect of the slender loops (7mm) with a slight intraperitoneal effusion.
Jejunal biopsy showed Jejunal mucosa bristling with villi of normal height and lined by a regular columnar epithelium, normosecretant. The glands are well differentiated nomosecretantes. The villus axis is congestive edematous with lesions of lymphangiectasia. The chorion harbors an inflammatory lymphoplasmacytic infiltrate mixed with a few neutrophils (Figure 2).
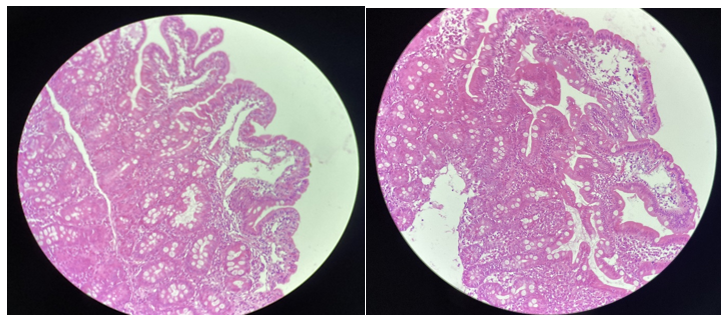
Secondary causes are ruled out before normal echocardiography, absence of lymphatic obstruction on abdominal ultrasound, normal fecal calprotectin, and negative ASCA and ANCA. The karyotype was normal ruling out Turner syndrome.
The diagnosis of Waldmann's disease was retained in our patient, she received albumin infusions and high protein diet, poor in long-chain fatty acids, enriched in medium-chain fatty acids with supplementation of fat-soluble vitamins and iron.
The evolution was marked by the stabilization of his clinical condition.
Primary intestinal lymphangiectasias or Waldmann's disease were described in 1961 by Waldmann in 18 patients [5], they are due to lymphatic dilations with lymph leakage into the intestinal lumen. It is a disease usually appearing in childhood [6], sometimes with growth retardation in severe forms and of which there are some familial forms [7].
The secondary etiologies of intestinal lymphangiesctasia must be ruled out in order to retain Waldmann disease.
In children, the disease is usually diagnosed before the age of 3, sometimes with very extensive and fatal forms [5, 8]. The main clinical signs are: edema, diarrhea, ascites, asthenia, abdominal pain, nausea, vomiting, weight loss or lack of weight gain, growth retardation. Malabsorption can lead to a deficiency in fat-soluble vitamins (A, D, E, K ), episodes of hypocalcemia sometimes responsible for convulsions [5]. The association with celiac disease has been occasionally reported in children. The degree of severity depends on the anatomical location and the extent of the lymphatic abnormality [9]. While edemas in Waldmann's disease are frequent and associated with hypoproteinemia, lymphoedemas are rarer but can affect the lower and upper limbs. In the course of the disease, they usually occur after gastrointestinal involvement which is symptomatic and prominent [6].
Intestinal lymphangiectasias are recognized during oesogastroduodenal fibroscopy, the suggestive macroscopic appearance corresponds to lymphatic dilations of yellow / brown color (chylomicrons) of variable density and size. Duodenal biopsies confirm the diagnosis with the demonstration of lymphatic dilations, more or less important. The biopsies must be multiple because the lesions can be very localized. There is no villous atrophy or inflammatory infiltrate [10] [11].
The other morphological examinations are of little value. The thoraco-abdomino-pelvic CT scan can show dilation of the slender loops and a thickened aspect of the mucous folds [11]. Indirect laboratory abnormalities are suggestive of LIP: hypoproteinemia, hypo albuminemia, hypogammaglobulinemia (drop in Ig G, A, M) or lymphopenia [12].
The primitive nature of intestinal lymphangiectasias can only be affirmed after having eliminated the secondary causes of digestive lymph loss: malformations of the lymphatic channels, diseases of the lymph nodes (lymphoma, metastases, tuberculosis, filariasis, Whipple's disease), compression of the ducts lymphatics (tumors, retroperitoneal fibrosis, mediastinitis), heart disease with venous pressure, thrombosis of the inferior vena cava, lympho-intestinal fistulas, iatrogenic lesions of the lymphatic system (lymph node excision, radiotherapy) [6].
The treatment is based on a diet low in long chain fatty acids, to limit the pressure in the intestinal lymphatics and prevent their rupture in the digestive lumen. The diet is supplemented with medium chain fatty acids, which are absorbed directly in the portal circulation. Oral supplementation with fat-soluble vitamins (especially D) is essential [12], it is sometimes useful to treat iron deficiency (oral or parenteral). Diuretics may sometimes be necessary in persistent and disabling edema. The treatment generally allows the regression of the laboratory abnormalities but in some cases there may be biological stigmata of lymphatic leakage while the clinical signs may disappear if the diet is strict [13]. Treatment with subcutaneous octreotide has also been reported in a few cases where clinical signs persisted with diet [14] [15]. For lymphedema, it is not sensitive to the fat-free diet but should be treated with standard compression-compression techniques [16].
Primary intestinal lymphangiectasia or Waldmann's disease is a chronic disease requiring a strict, prolonged and restrictive hypo-lipid diet, supplemented with medium-chain triglycerides and fat-soluble vitamins.
In pediatrics, it should be suspected when an infant develops chronic diarrhea: malnutrition, edema, hypoalbuminemia and lymphopenia in the first months of life. The quality of life of these patients is often impaired by the presence of edema and / or lymphedema and frequent asthenia. Early diagnosis and appropriate nutritional treatment corrects the loss of lymphocytes, immunoglobulins, proteins, fats and micronutrients which can lead to repeated infections and, in some cases, death.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
