
Review Article | DOI: https://doi.org/10.31579/2690-1919/515
1Rheumatologie im Bavariahaus, Bavaria Health Center, Augustenstr. 10, 80333 Munich, Germany.
2Institute of Rheumatology, Na Slupi 4, Prague 2, Czech Republic.
3Internistisches Klinikum München Süd, Am Isarkanal 36, 81379 Munich, Germany.
4 Radiologie München, Säbener Str. 51, 81547 Munich, Germany.
5Retired, Institute of Physiological Chemistry, Philipps-University Marburg, Karl-von-Frisch-Str.1, 35033 Marburg, Germany.
*Corresponding Author: Werner Löffler, Rheumatologie im Bavariahaus, Bavaria Health Center, Augustenstr. 10, 80333 Munich, Germany.
Citation: Werner Löffler, Blanka Stibůrková, Manfred Gross, Martin G. Mack, Monika Löffler (2025), Treat to Target Suggestions in Gout - a Different View. J Clinical Research and Reports, 19(3); DOI:10.31579/2690-1919/515
Copyright: © 2025, Werner Löffler. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 05 March 2025 | Accepted: 24 March 2025 | Published: 01 April 2025
Keywords: gout; uric acid lowering treatment; hypouricemia; treat to target; chronic inflammation; mortality; cardiovascular risk
Despite significant progress has been made with understanding and treating gout since the mid-20th century, the disease remains challenging to manage to for a considerable percentage of patients. Treatment success often requires several years to achieve.
We aim to demonstrate that successful gout treatment can be accomplished much earlier by use of moderate doses of currently available medications. Compared to pegloticase, regimens described here will be similarly effective in a minority of patients and only slightly less effective in a high percentage of patients. By employing such treatment strategies, gout can be cured within one to two years in most patients, with treatment durations exceeding two years being rare.
Given the long-term consequences of chronic subclinical inflammation associated with gout, we conclude that the disease can be cured very much earlier than suggested by current guidelines.
Gout is a highly prevalent metabolic disease characterized by inflammatory sequelae originating from mono-sodium urate (MSU) crystal deposits, which occurs after longterm persistence of increased levels of uric acid in blood and tissues. Since MSU crystals can be dissolved by reduction of serum uric acid (sUA) levels, gout is a curable disease. The rising incidence of gout significantly affects individual health and presents substantial economic burdens, including reduced productivity and increased healthcare costs [1-3].
Treat To Target (T2T) recommendations have been established for the treatment of gout, similar to strategies used in a variety of chronic diseases including hypertension, metabolic, and rheumatic diseases, and it has been suggested to use a Gout Activity Score for this purpose [26].
This manuscript discusses various aspects of defining treatment targets and strategies for achieving them in uric acid lowering treatment (ULT).
The Treat-to-Target (T2T) approach involves implementing therapeutic measures to achieve a final state where the patient is free of symptoms or signs related to the disease. In gout, this means that sUA levels must normalize and all symptoms and signs attributable to UA crystal deposition must resolve. T2T recommendations were originally established for managing chronic diseases such as hypertension, metabolic disorders and rheumatic diseases. More recently this approach has also been applied to gout treatment (reviewed in [4]). Exept for acute flares, however, treating gout is not comparable to the treatment of chronic inflammatory diseases.
Clinical studies on UA lowering drugs (ULDs) in gout typically assess efficacy after six and twelve months [5, 6] or longer. The sUA concentrations measured at these time-points are called primary end points. While this approach is valuable for clinical studies (to determine how many patients have reached a certain sUA concentration after a certain time), it does not necessarily reflect the true UA lowering effects of a given drug at its steady-state dose.
In contrast to the treatment of inflammatory diseases – where immunosuppressive agents take weeks to reach a steady-state concentration, followed by a gradual increase in efficacy – ULDs reach a steady-state serum concentration within one to two weeks. At this time-point, the maximum achievable sUA reduction has been attained and remains stable, while the dissolution of MSU crystals becomes a passive process.
Studies have demonstrated that the rate of tophus dissolution depends on the degree of undersaturation in surrounding tissues, which is directly related to the extent of sUA reduction [7], following the general principle that substances present in adjacent compartments at different concentrations will diffuse accordingly [8]. This concept has been compared to a flooded meadow – where water recedes faster as the adjacent water level drops. Clinical observations align with this model, showing that crystals dissolve twice as fast at sUA levels below 4 mg/dL compared to levels above 5 mg/dL [9].
Moreover, the time required for complete crystal dissolution is directly determined by the sUA target set at treatment initiation [10]. Observations in patients receiving uricase therapy further support this principle. In persistent responders, tophi have been shown to dissolve rapidly within short periods [11-14]. Given this evidence, there is no rationale for requiring additional long-term studies to confirm the efficacy of rapid sUA reduction.
The sUA concentration seen immediately after a steady state of the drug concentrations has been reached, may slowly decline further, during constant doses of the drugs, indicating ongoing dissolution of tophi. A final, constant sUA, in equilibrium with interstitial fluids, will be reached after complete dissolution of crystal depositions. Increased sUA concentrations at 12 months compared to 6 months, during controlled studies, at identical drug dosage, are likely due to lifestyle changes or increasing rates of non-adherence [5,15].
Current guidelines have recommend targeting sUA levels below 6.0 mg/dl [16] or below 5 mg/dl [17], a later recommendation reducing sUA below 6.0 mg/ or below 5.0 mg/dL [18], with a target of below 5.0 mg/dL specifically for patients with severe gout [19]. No additional target thresholds have been proposed in the past two decades [20]. While these limits may be adequate for patients with a low burden of tophi, they may be insufficient for those with severe tophaceous gout.
Patients following standard guidelines may experience little to no improvement for several months, potentially leading to frustration and poor adherence. This effect is similar to the increased frequency of gout flares observed after initiating ULT. In patients with a high crystal burden, the slow dissolution process may reinforce the misconception that gout is an untreatable chronic disease – both among patients and healthcare providers [21]. Additionally, defining "severe gout" based on estimated crystal deposits remains challenging.
Numerous factors contribute to the highly variable presentation of gouty flares [22]. However, there is no evidence that different ULDs induce flares via distinct mechanisms. The primary trigger for flares during early ULT is the dissolution of MSU crystal deposits, although additional modifying factors may play a minor role. Patients should be informed in advance that increased flare frequency during the early phase of treatment indicates successful sUA reduction and crystal dissolution. Frequently follow-up visits during this period allow for regular patient counselling, minimizing the risk of non-adherence. Importantly, flare frequency should not be classified as an adverse event or included in the safety assessment of ULDs [5, 6, 23]. Instead, higher flare occurrence is an indicator of treatment efficacy and successful sUA lowering [24].
The number of flares has been shown to be lower during the first year of ULT, if increasing the dose gradually within 6 months, compared to immediate full-dose initiation [25]. However, gradual dose escalation results in slower crystal dissolution and may extend the duration of flare susceptibility. Notably, no studies have examined whether a slower dose escalation reduces the total number or severity of flares or simply redistributes them over a longer period.
Some T2T strategies for gout have incorporated composite disease activity scores, such as the Gout Activity Score [26], to monitor treatment response. Unlike inflammatory diseases – where disease activity scores reflect systemic immune activation – T2T strategies in gout should primarily focus on biochemical targets, specifically sUA reduction [4, 27]. We argue that sUA reduction should not be considered just one target among many, but rather the primary determinant of treatment success. Early and aggressive sUA reduction is crucial, although potential risks, such as severe adverse effects when initiating full-dose allopurinol, should be considered [28].
A clear distinction must be made between clinical targets and sUA targets. Normalizing the sUA is the foundation of gout treatment, rendering all other targets secondary. The serum level of UA - the key factor determining treatment success – should not be considered just one variable among many. Instead, it must be maintained at a normal or subnormal level consistently from the very beginning of treatment. Including the sUA into a composite disease activity score means to include uncertainties, which should have been ruled out at the start of long-term ULT. If hyperuricemia persist beyond six to eight weeks of ULT initiation, the treatment target has been missed, and clinical improvements cannot be expected within a reasonable timeframe.
Before reaching the clinical target, in the case of flares, symptoms should be treated symptomatically, and their occurrence may prompt further sUA reduction. Additionally, non-gout-related symptoms and signs may mimic acute flares. We do not see, how a disease activity score could be helpful in such a situation. Notably, gout activity scores have not been included in recent guideline suggestions.
Studies following guideline-recommended procedures have shown that ultrasound signs of gout, although reduced, remained detectable after 1 year [29], or after 2 years of ULT [30]. Similarly, crystal volumes measured by DECT decreased by 28% only after two years [31], indicating that chronic subclinical inflammation caused by MSU crystal deposits had not been fully resolved. Accordingly, a prospective study showed that ULT significantly reduced the risk of all-cause, or cardio-vascular mortality after 6.5 years [32]. No mortality risk reduction was observed after 2 years [32], and another study found no significant benefit after 5.25 years [33]. Considering the exponential nature of crystal dissolution, these findings suggest that full tophus resolution may take a decade or longer under current guidelines. Indeed, some studies have documented persistent tophi even after ten to twelve years of treatment [34, 35].
Regardless of measured sUA levels, the regression of tophi and the progression of articular damage must be closely monitored. In rare cases, rapid dissolution of bony tophi can lead to inadequate reossification resulting in mutilating arthritis [36]. However, re-ossification generally occurs slowly, likely resembling the process of fracture healing rather than soft tissue healing.
One of the most intriguing unsolved questions in gout is why some patients develop MSU crystal deposits and clinical gout, while others, with identical levels and duration of hyperuricemia remain asymptomatic. In terms of treatment, however, there is no doubt that we have the tools to cure gout.
Both patients and physicians should be made aware that gout is fundamentally a treatable disease, provided two essential principles are followed: first of all, gout can be completely cured, if sUA levels are maintained within or below the normal range for a sufficient period. Second of all, the speed of MSU crystal dissolution is directly proportional to the degree of sUA reduction – meaning the lower the sUA the faster the resolution. And a higher frequency and/or severity of gout flares during the initial treatment is a sign of effective urate lowering, not treatment failure.
Non-adherence
A third critical factor– presumably the most important – is the problem of adherence. In case the expected treatment success is not achieved, non-adherence should be ruled out. During the last decades, whenever numbers of patients were mentioned in publications not reaching any treatment target, typically no information was provided about investigating possible causes [5]. Instead, non-response, or insufficient response to the prescribed drug was generally assumed to be the reason.
It has been demonstrated, however, during controlled studies, that close to 40% of patients – initially classified as non-responders to standard medication – achieved normal sUA levels when receiving their previous medication under double-blind regimen [5]. A systematic review of adherence to allopurinol treatment for gout reported that 57% of patients did not persist on allopurinol treatment after one year and 77 percentage after five years of starting treatment [37]. Poor adherence remains a major barrier to effective gout treatment and can lead to more severe disease and increased gout-related healthcare costs.
A large cohort study using data from the United States Veterans Affairs national databases identified several factors associated with higher adherence to allopurinol, including older age, greater comorbidity burden, higher BMI and higher likelihood of receiving care in a community outpatient clinic or rural healthcare facility [38]. Implementing nurse-led care – which included patient education and active engagement - dramatically improved adherence, with 95 % of patients reaching target sUA levels compared to only 30% under standard care [39].
Prior to the 2000s, it was well-known that non-compliance can be investigated by tests such as measuring allopurinol and oxipurinol in serum, or urine, and assessing xanthine oxidase-inhibition by quantifying hypoxanthine and xanthine, which would be elevated compared to physiological concentrations. Nowadays, laboratories offering these tests in routine care – including university medical centers - are rare because of the low numbers ordered. Samples sent to specialized laboratories for investigating suspected biochemical causes of insufficient treatment response dropped from two dozen per year to below one per year between 2015 and 2019 (Purine Laboratory, Guy´s and St. Thomas´ Hospital, London, L. Fairbanks, 2021; personal communication). In total, we are aware of just three patients with an abnormal response to allopurinol having been documented [15], while no such data have been reported with other ULDs ever.
Oxypurinol, the active metabolite of allopurinol, is primarily excreted unchanged via the kidneys. The measurement of plasma [40], or urinary oxypurinol concentrations [41] has been shown to identify low adherence to allopurinol in clinical trials and may be a more reliable tool to asses adherence than pill counts. Indeed, due to the short plasma half-life of allopurinol versus oxypurinol, it is possible to definitely confirm, wether or not a patient had taken allopurinol on a certain day, if measuring both parameters within few hours after breakfast.
Incorporating urinary oxypurinol assessment into routine clinical practice could have several advantages. It provides an objective and quantifiable measure of adherence that enables timely intervention and additional education for non-adherent patients, which could improve adherence and overall gout management and prevent disease exacerbations and complications associated with uncontrolled hyperuricemia. Moreover, in comparison to serum oxypurinol, urine collection is a non-invasive procedure and can be repeated multiple times on different days to determine partial adherence. In clinical practice, it would be useful and cost-effective to measure oxypurinol in poor responders to allopurinol therapy before switching to the more expensive therapy with febuxostat.
Bone erosions
The positive effects of ULT have been demonstrated by numerous clinical studies. In the two years study mentioned above, standard treatment (sUA, <0.36 mmol/l, or 6 mg/dl) resulted in a 28% reduction of tophus size [31]. When comparing standard treatment (sUA, <0,3 mmol/l, or 5 mg/dl) to more intensive treatment (sUA, <0.2 mmol/l, or 3,36 mg/dl) [42], a significant number of patients did not reach the sUA target in both groups, and small increases in bone erosion scorses were observed in both groups. It was concluded that, compared to standard treatment, more intensive ULT was not superior, was difficult to achieve with oral ULT, imposed a higher medication burden, and did not improve bone erosions scores in erosive gout.
We believe that these conclusions do not align with the actual findings. If a treatment regimen results in 28% reduction in total tophus burden within two years, the individual size of tophi present may be reduced, the number of erosions free of MSU crystals, i.e., free of inflammation, however, most likely will be close to zero. Moreover, bone erosions cannot be expected to improve until the local inflammation is stopped, i.e., the last MSU crystals being dissolved. Hence, during the first years of treatment, if following guideline recommendations, we cannot expect statistical analyses to show a significant improvement in inflammation-related outcomes parallel to the reduction in tophus size – much like the lack of improvement in mortality rates during the initial years of treatment. We propose that a significant difference will become evident, if such studies are extended over longer periods. That said, with very intensive treatment (sUA <1 mg/dL), similar results can be achieved in a much shorter time, as demonstrated with pegloticase treatment [13].
As previously demonstrated, zoledronate was not effective in preventing bone erosions caused by MSU crystal deposits [43], and denosumab did not result in improvement in bone erosion scores in patients with erosive gout [44]. However, studies have also shown that bone erosions gradually improve after complete dissolution of MSU crystals, even when ULT is discontinued, and sUA levels exceed 9 mg/dL [11]. This demonstrates that it is the combination of crystals and inflammation, rather than hyperuricemia that causes the problem. It also supports the conclusion that hyperuricemia itself is not the cause of inflammation.
Gout, premature mortality, and correlations to other chronic diseases
A recent study has again raised awareness of the increasaed risk of cardiovascular disease (CVD) in patients with gout [45]. A significant association was found between gout and several CVDs, with a hazard ratio [HR] 1·58 [95% CI 1·52–1·63], and between each of the 12 CVD considered. Gout was also linked to a higher likelihood of hospitalization for CVD (HR 1·33 [95% CI 1·28–1·38]) and CVD-related mortality (HR 1·41 [1·24–1·60]). These findings highlight the importance of targeted screening and management of CVD risk factors in gout patients.
Previous studies have shown that during ULT according to guidelines, it takes at least 6.5 years before premature mortality rates begin to improve significantly [32]. Due to the exponential nature of crystal dissolution, complete clearance of deposits and resolution of inflammation can only be expected to occur several years later. Consequently, treating gout according to current guidelines still allows persistent inflammation to exist for at least ten years. If we also consider the period between the initial deposits of MSU crystals and the start of ULT, this timeframe extends to a minimum of twelve to 15 years of chronic inflammation, causing damage to the cardiovascular system and other organs.
A population-based study conducted between 1999 and 2014, during which treatment guidelines were already in use, found no reduction in premature mortality among patients with gout [46]. Similarly, a previously cited study [33] reported no reduction in CV risks during a 5.25-year period of allopurinol treatment. However, when comparing doses of 100 mg/d versus 300 mg/d or higher, a reduction in risk was observed in the higher-dose group. Other studies [47] have reported similar findings, reinforcing the notion that achieving treatment targets depends not only on the extent of sUA reduction but also on the duration of treatment.
Over many years, a number of chronic diseases has been demonstrated to be correlated to gout, whereas studies on patients with so-called asymptomatic hyperuricemia have yielded contradictary results [48]. The term “asympomatic hyperuricemia” has traditionally been used to describe cases in which no acute gout attacks have occurred. However, with the introduction of ultra-sonography and DECT, a significant number of “asymptomatic” patients have been found to have crystal deposits, placing them at risk for subclinical chronic inflammation. The reduction of ultrasound-detected MSU crystal deposits has been shown to correlate with treatment success [49], and ultrasound has been proposed as a tool to identify MSU crystal deposits as a basis for determining wether treatment should be initiated [50].
It has been stated that the extent to which the sUA reduction can reliably lower CV risks remains uncertain [51]. However, this is not merely a matter of the degree of reduction over time. The most important point will be whether complete dissolution of MSU crystal deposits is achieved, thereby eliminating inflammation and other associated risks. Clinical gout should therefore be defined as the combination of hyperuricemia and confirmed MSU crystal deposits, regardless of whether acute attacks have occurred.
Currently, it remains unclear whether the occurrence of acute attacks is simply a matter of time or whether some patients remain asymptomatic despite confirmed crystal deposits on imaging. Additionally, there are no clear guidelines on how to manage patients with documented crystal deposits who have never experienced acute gout attacks. It is reasonable to assume that crystal deposits will continue to grow necessitating UA lowering treatment irrespective of acute attacks.
How to Approach the Target
After intensive discussion and information on the different ULT approaches, patients should be given the opportunity to decide for themselves whether they prefer an intensive treatment course of ULT – with potentially more frequent and severe attacks – or a more moderate approach. In our experience, numbers and intensity of attacks are variable to a large extend not only with moderate (sUA, <4.0 mg/dl), but also with intensive treatment (sUA, <2 mg/dl), as demonstrated by the two cases following.
Patient 1 (Figure 1) had witnessed his father suffering from recurrent gout attacks for many years while on UA lowering medication (possible with partial adherence). From his early twenties, he experienced approximately four attacks per year for a decade, and six attacks per year in his mid thirties. At his first visit at age 37, after discussing different strategies, he opted for an intensive protocol (Figure 1). His regimen included febuxostat, titrated from 40 mg to 80 mg and then 120 mg per day over two to three weeks at each step. Benzbromarone was added at doses of 25 mg, 50 mg and eventually 100 mg per day, until his sUA dropped below 2.0 mg/dL. Between weeks 4 and 23 of ULT, acute attacks occurred with increasing frequency, peeking at three per month, with a final attack occurring six weeks later. Instead of colchicine or other prophylactic medication, he took prednisolone as needed – 20 or 30 mg for one or two days, followed by 5.0 mg for up to one week – or in some cases, without additional intermittent doses, depending on the severity and duration of his attacks. His sUA remained below 4 mg/dL after three weeks of treatment and below 2 mg/dL between weeks 23 until 46 weeks. Over the following six months, his average sUA stabilized at 3.3 mg/dL. He then continued on febuxostat 80 mg per day, with an average sUA of 4.6 mg/dL. A constantly normal CRP level was achieved after 58 weeks.
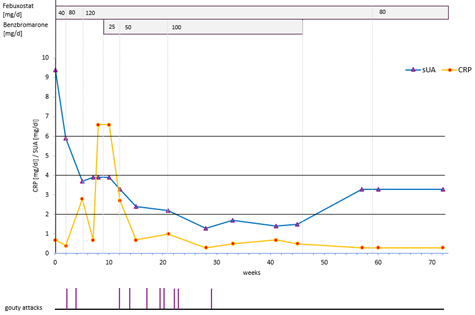
Figure 1: Intensive uric acid lowering treatment in gout (target serum uric acid,<2,0 mg/dl), Patient 1. (| | gouty attacks).
In this patient, increasing the dose of febuxostat from 80 to 120 mg/d did not lead to a further reduction in sUA levels (80 mg, 3.7 mg/dl; 120 mg/d, 3.9 mg/dl, 3.9 mg/dl), what has been described previously [52]. Similarly, after discontinuation of benzbromarone, the sUA remained at 3.3 mg/dl for both febuxostat doses, further illustrating that lower sUA levels were achieved at identical doses after complete crystal dissolution.
Patient 2 (Figure 2), 72 years, had had gouty attacks from the age of 60. He had started taking allopurinol, 300 mg/d, followed by an acute oligo-articular attack lasting three weeks by the time of his first visit. Treatment was switched to febuxostat, 80 mg/d, and increased to 120 mg/d 6 weeks later. His attack had subsided after a total of 6 weeks (3 weeks after switching to febuxostat). Another oligo-articular, migratory attack of 5 weeks duration started 8 weeks later. On a visual analogue scale, he rated his pain from 3 to 7, and he took ibuprofen as needed with a maximum of 3x 600 mg/d. Despite the presence of large crystal volumes (Figure 3; first DECT done 4 weeks after start of ULT; last attack ending 12 weeks after first DECT), no further acute attacks occurred. Throughout the course of combined treatment, the sUA levels fluctuated (Figure 2). Obviously, the patient was willing to co-operate, but frequently forgot to take single tablets, or took two when supposed to take one only, or did not ask for prescriptions in time. Nonetheless, his sUA remained in the low-normal to hypouricemic range and treatment success was as expected during an intensive course. A follow-up DECT taken 54 weeks after the first showed a single small deposit remaining in the lateral compartment of his left knee, not detectable by additonal imaging 2 years after start of treatment. After 3 years, the patient continued on allopurinol, 300 mg/d (sUA, 3.6 mg/dl).
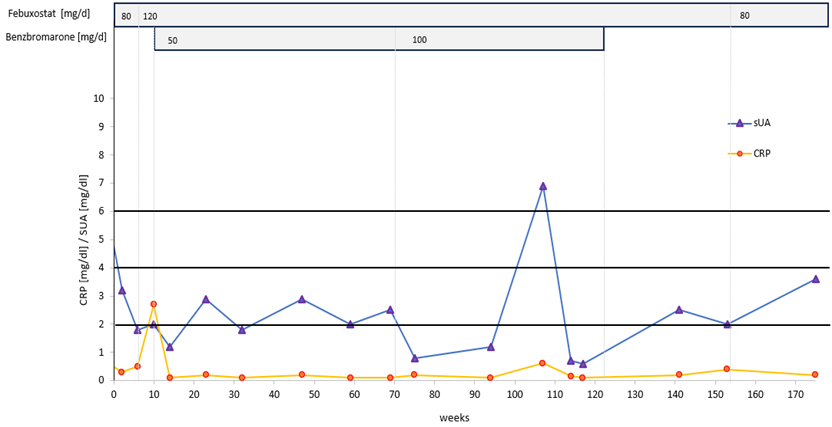
Figure 2: Intensive uric acid lowering treatment in gout (target serum uric acid, <2,0 mg/dl), Patient 2
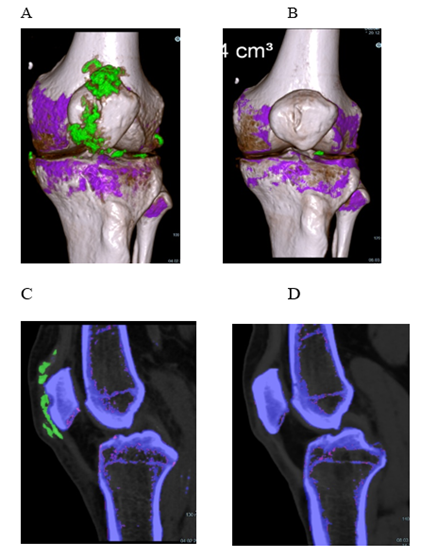
Figure 3: Patient 2, dual energy computed tomography of his left knee. 3D reconstruction (A+B) and sagittal reformatted source images (C+D). – A, C and B, D: 4 weeks and 61 weeks, respectively, after start of uric acid lowering treatment
During intensive ULT, progress will be fast enough not to necessitate regular monitoring to adjust targets according to stages based on severity [4]. Clinical symptoms should be managed individually, while sUA levels are maintained in the low range, until any symptoms or signs related to gout have completely subsided. It does not make sense to follow remission, with the dose of the ULD being kept constant and the sUA remaining in the mid to upper normal range, waiting for clinical remission to be achieved over the course of many years. We consider this a needless delay of the treatment success intended, and presumably will lead patients to be non-adherent.
Contrary to previous claims, the dissolution of tophi does not necessarily take years with currently available ULDs [11]. While it has been suggested that profound sUA reductions comparable to treatment with pegloticase are not possible with oral ULT [13], pegloticase is not the only viable solution. Similar outcomes can be achieved using standard medications.
Hypouricemia – Is It a Risk?
Inborn errors in metabolism associated with hypouricemia can be divided into two groups: hypouricemia due to lack of UA synthesis (xanthinuria) and hypouricemia due to increased renal UA excretion (renal hypouricemia, RHUC). As stated previously, apart from urolithiasis in both xanthinuria and RHUC, or exercise induced acute renal failure in HRUC [53, 54], problems associated with a low sUA are unknown [55]. During the last decades, however, it was found that in xanthinuria, in addition to the radiolucent xanthine stones occurring in 40% of patients, xanthine crystal deposition in soft tissues developing over a longer period of time will result in arthropathy, myopathy and duodenal ulcers in 10% of patients. The most common of them is myopathy, with muscle pain and cramps being typical symptoms frequently precipitated by strenous exercise [56].
Unlike xanthinuria, RHUC has not been linked to symptoms outside the kidneys and urinary tract. However, compared to healthy individuals, the prevalence of renal stones is 6-7 times higher in patients with RHUC. Despite a substantial reduction of the strong antioxidant effect ascribed to UA, patients with both kinds of hypouricemia generally remain asymptomatic aside from the described symptoms [57].
One of the proposed factors contributing to the renal failure occurring in RHUC is impaired flow-mediated arterial dilation due to low sUA levels [58-60], leading to vasoconstriction and renal ischemia. Additionally, significantly lower blood hypoxanthine levels and increased urinary hypoxanthine excretion have been observed in RHUC patients after exercise, potentially resulting in ATP loss in renal tubules and subsequent renal damage [61]. Muscle ATP degradation during exercise may surpass salvage capacities, further increasing UA production [54]. Oxidative stress markers did not differ between healthy subjects and patients with RHUC before and after exercise, and no effect of uric acid as a radical scavenger was observed [61], while others concluded hypouricemia to result in loss of antioxidant capacity [60].
In the study by Sugihara [58], flow-mediated dilation was found only in those patients with sUA <0.8 mg/dl, but not in hypouricemic patients with somewhat higher levels, suggesting genetic factors to be the cause, but not low sUA concentrations [59]. Symptoms similar to exercise induced renal failure have never been reported in patients with xanthinuria [53], reinforcing the notion that low sUA concentrations cannot be the decisive factor with this respect.
In the context of exercise induced renal failure, renal overexcretion of UA been mentioned very rarely. In contrast to multiple statements after the millenium, generally reporting of renal excretion being around two thirds to 80 percent of total excretion [62-65], previous studies suggested that the proportion of renal excretion will follow renal UA clearance [54, 66-69]. It has been stated previously that the hyperuricosuria seen in patients with HRUC probably reflected diversion of intestinal UA elimination to urinary UA excretion, and there was no evidence in the hypouricemic hyperuricosuric subjects for purine overproduction [54].
We can expect, therefore, in RHUC, above 90% of total UA production to be excreted renally. For instance, assuming UA excretion to be 600 and 300 mg/d via kidneys and gut, respectively, in healthy males during an unrestricted diet, and comparing these to a patient with RHUC, with similar body composition and diet, this would mean that renal excretion will be around 800 to 900 mg/d, while intestinal excretion will be <100 mg/d, lifelong. Renal UA clearances, or fractional clearances, as high as 173 ml/min, or 1.69, respectively, have been reported [54].
It is tempting to speculate that in RHUC disturbances of arterial mechanisms will remain asymptomatic during normal conditions, but may contribute to renal damag whenever additional factors will occur, such as an increase in UA excretion (degradation of ATP) or reduced pH of urine, or fluid loss/increased concentration of urine in the context of exercise. No such risks exist with intensive ULT. During febuxostat, 80 mg/d, renal excretion of UA will not exceed 50% of control values [52]. Even if high-dose uricosurics were added, shifting intestinal excretion towards renal elimination, total renal UA excretion would still remain within normal limits.
In 1981, it was postulated that urate functions as a physiologically significant free radical scavenger [70], prompting concerns that hypouricemia might be a risk factor for neurodegenerative and other diseases. Some studies suggested urate may provide protection against multiple sclerosis and Parkinson´s disease [71-75]. In contrast, higher-than-normal sUA levels have been linked to dementia and cognitive decline [76], with varying findings regarding UA metabolism in Alzheimer, Parkinson and multiple sclerosis [77]. Recent studies have suggested that low sUA levels may also be associated with increased risks for cancer [78], postoperative acute kidney injury [79], decline in kidney function in healthy individuals [80] and higher total and cardiovascular mortality [81, 82].
There have been concerns that the low sUA produced by treatment with pegloticase might increase H2O2 to toxic levels [83]. However, during treatment with pegloticase, in patients with constantly low sUA values (0.9 +- 0.5 mg/dl), F2-isoprostane levels, known to be associated with oxidative stress, have not been found to be elevated. Moreover, maintaining sUA below 1.0 mg/dl for 15 weeks was associated with a trend towards a decline in F2-isoprostan levels from baseline. Additionally, no increased H2O2 production was observed in vitro in the blood of these patients [84] and oxidative stress markers remained unchanged in healthy individuals and RHUC patients pre- and post-exercise [66].
From studies mentioned above, the conclusion was drawn that the sUA should not be reduced <3 mg/dl in the long term, that is, not for several years [19]. However, none of the factors found to be associated with hypouricemia in population studies or experimental clinical studies, have been confirmed to be clinically relevant by prospective controlled studies. Furthermore, none of these risks have ever been described in patients with xanthinuria, or renal hypouricema, or in women having had hypouricemia in the premenopausal state, which means, for decades.
If future studies confirm significant risks associated with transient hypouricemia during early intensive ULT, the magnitude of this risk is likely negligible compared to the well-documented dangers of chronic inflammation due to crystal deposits, which persists for many years when sUA is maintained at only below 5.0 or below 6.0 mg/dL.
It has been concluded previously that maintaining the sUA near or below 2 mg/dl would probably be safe, and the potential benefit to patients with severe gout would justify taking hypothetical risks [85]. Assuming a significant risk also being associated with short periods of hypouricemia, this would render pegloticase treatment obsolete in clinical medicine, which has been followed, with clinical studies, for up to 30 months [86].
How to Proceed in Daily Care
During early ULT, it is practical to define a time period where the inital low target sUA is maintained beyond the last flare. This should also consider normalization of inflammatory markers, and/or dissolution of the last documented tophus based on clinical, laboratory, or imaging results. We recommend extending this period until the time from starting ULT to the last flare will be doubled, inflammation markers normalize, or the last tophus dissolves. This should be followed by a period with the sUA being kept in the low normal range (<4 mg/dl), lasting as long as the period preceding to make sure that all MSU crystal depositions being dissolved.
In case of intensive treatment, this would mean, for example, to remain at a sUA <2.0 mg/dl for one year, if the last flare had occurred after 6 months, and thereafter keeping the sUA at <4 mg/dl for another year, followed by the final target at <6 mg/dl. From the DECT investigations done in Patient 2, it would appear that, also in the case of high tophus volumes, a sUA <2.0 mg/dl will be sufficient to dissolve all MSU crystal depositions within 2 years in most cases. In the case of moderate ULT (<4 mg/dl), this could mean, for example, to remain at a sUA <4 mg/dl for 4 years, if the last flare having occurred after 2 years. Imaging studies might be adequate in this case to define the time of continuing at this sUA level. With intensive ULT, we do not think it wise to use single drugs in high dosages. As shown in Table 1, increasing doses beyond moderate levels provides only marginal additional benefits while significantly increasing the risks of adverse effects.
| Serum Uric Acid | ||||
|---|---|---|---|---|
| Control (mg/dl) | Medication (mg/dl) | Medication (% of control) | ||
| Löffler and Gröbner [87] (healthy controls, n = 4; purine-free diet versus purine-free diet plus RNA, intraindividual comparison) | Allopurinol (mg/1.73 m²/d) | Purine-free formula diet | ||
| 250 | 3.4 ± 0.7 | 1.8 ± 0.4 | 52.9 | |
| 500 | 3.6 ± 0.6 | 1.6 ± 0.25 | 44.4 | |
| RNA, 4 g/d | ||||
| 250 | 6.9 ± 0.3 | 3.6 +- 0.3 | 52.2 | |
| 500 | 6.1 ± 1.25 | 2.5 ± 0.35 | 41.0 | |
| Khosravan et al. [52] (healthy controls, n = 10 each) | Febuxostat (mg/d) | |||
| 10 | 4.98 ± 0.82 | 3.64 ± 0.78 | 73.1 | |
| 20 | 4.83 ±1.14 | 3.21 ± 0.87 | 66.5 | |
| 30 | 4.24 ± 1.34 | 2.62 ± 0.87 | 61.8 | |
| 40 | 5.29 ± 1.77 | 3.22 ±1.31 | 60.9 | |
| 50 | 4.81 ± 1.34 | 2.59 ± 0.94 | 53.8 | |
| 70 | 4.43 ± 0.98 | 2.23 ± 0.87 | 50.3 | |
| 90 | 4.51 ± 1.06 | 1.78 ± 0.62 | 39.5 | |
| 120 | 4.66 ± 1.04 | 1.56 ± 0.36 | 33.5 | |
| 160 | 4.83 ± 1.14 | 1.44 ± 0.71 | 29.8 | |
| 180 | 5.26 ± 1.06 | 1.49 ± 0.51 | 28.3 | |
| 240 | 5.11 ± 1.12 | 1.23 ± 0.47 | 24.1 | |
| Schumacher et al. [88] (hyperuricemia, gout) | Febuxostat (mg/d) | |||
| 80 (n = 267) | 52 | |||
| 120 (n = 269) | 45 | |||
| 240 (n = 134) | 32 | |||
| Reinders et al. [89] (gout) | Allopurinol (mg/d) | |||
| 300 (n = 29) | 67 | |||
| 600 (n = 17) | 51 | |||
| Benzbromarone (mg/d) | ||||
| 100 (n = 22) | 58 | |||
| 200 (n = 7) | 54 | |||
Table 1: Experimental and clinical studies of uric acid lowering drugs, effects of dose increase. Löffler and Gröbner [87], absolute doses taken, 400 - 500, or 800 - 1000 mg/d; Schumacher et al. [88], Reinders et al. [89], absolute figures not reported.
If targeting the sUA concentration(s) suggested by guidelines, it would be difficult to define any timepoint, where patients can be assumed to be free of crystal deposits, i.e. free of the subclinical inflammation attributable to gout. Hence, we do not know from which timepoint on we can be sure that, whatever symptoms and signs will occur, gout can be excluded as an underlying cause a priori, and we do not know at which time it might be best doing imaging studies to confirm complete dissolution of crystal depositions. Therefore, more frequent appointments due to flares, or acute complaints not clearly attributable to gout, a higher number of diagnostic measures including invasive diagnostics and diagnostic imaging, may result in considerably higher costs compared to intensive treatment.
Compared to standard treatment, the intensive protocol outlined will not result in an inadequate total burden of medication. The short duration of higher numbers of drugs and dosages must be weighted against the many years of chronic inflammation that would otherwise persist. Additionally, chronic flare prophylaxis is unlikely to completely prevent the need 0f additional treatment in case of acute flares. We therefore suggest ensuring that patients are fully informed about the available treatment strategies and their implications.
In our experience, a minority of patients only opted for and adhered to an intensive treatment regimen, the most common reason being the intention to engage in particular future activities. Others decided to follow a moderate regimen, or, after experiencing an increased flare rate, decided not to further reduce their sUA. Notably, we are not aware of any patient deciding, after thorough discussion, to follow guideline suggestions from the start.
One could say that the necessity of T2T sUA has been existing from when researchers discovered that the symptoms of gout were produced by MSU crystals, hence, the target of treatment will be their dissolution. It is time now to stop discussing what was its evidence, or if we should aim for rapid crystal dissolution. It is difficult to understand, what else should ´treat to serum uric acid target` mean than reducing the sUA as low as necessary. With a T2T strategy, a lower gout flare rate cannot be named a desirable outcome [90]. Moreover, if we were using drug dosages resulting, after two years of treatment, in a persistent trend towards resolution of tophi and reduction in the rate of flairs [91], we clearly are going without the tools we have. Gout has long been a disease of high interest for rheumatologists and clinical immunologists [92]. However, studying inflammation and immunological mechanisms will not help with the decisive step necessary for curing gout, the re-direction of metabolic pathways resulting in dissolution of MSU crystal deposits.
We propose that the combined oral treatment described will be somewhat less effective compared to persistent responders to pegloticase on average, but can achieve similar efficacy in individual patients (120 mg/d febuxostat plus benzbromarone 100 mg/d; Patient 2, sUA <1.0 mg/dl). Compared to partial response to pegloticase, this oral ULT aproach offers superior outcomes, very much lower costs and significantly fewer adverse events – without the need of additional prophylactic immunosuppressive treatment to prevent anti-drug antibody reactions.
The increasing prevalence of gout worldwide and the rising number of hospital admissions due to acute flares, reinforce the urgent need for aggressive, target-driven therapy in this highly treatable disease [1]. It should not be standard practice to merely reduce sUA to below 6 mg/dL, knowing that this approach will postpone reaching the therapeutical target until the last possible moment. Meanwhile, patients continue to accumulate the negative consequences of MSU crystal deposition and chronic inflammation over years.
Based on current knowledge, any chronic disease associated with chronic inflammation will likely correlate with gout, if both conditions have coexisted long enough. Rather than continually identifying new diseases that correlate with gout, we should focus on treating gout to the point where all crystal deposits have dissolved. Only then can we meaningfully compare gout to other chronic inflammatory diseases – at which point, the assumed correlation may no longer exist, and the prognosis of patients with gout may align with that of the general population, as was proposed decades ago [93].
Current guideline recommendations of sUA targets below 6.0 mg/dL or below 5.0 mg/dL, are ultimately based on expert opinion, derived from various observations. In daily care, these thresholds have been followed too rigidly. Instead, guidelines should be understood as allowing flexibility in applying any sUA target below 6.0 mg/dL, depending on the patient´s individual situation.
By following the intensive protocol outlined, the majority of gout patients with low amounts of MSU crystal deposits, could be cured within one year. For patients with higher tophus volumes or those on a moderate regimen, this timeframe extends to two years or in rare cases, up to three years. Importantly, this can be achieved using standard medications at moderate doses, and with acute attacks resolving much earlier in the course of treatment.
Given the high rate of adherence issues among gout patients, we do not believe it is beneficial to burden them with discussions about lifestyle modifications and dietary restrictions in the early phase of ULT. These aspects should certainly be addressed, but only after acute attacks have ceased.
Unlike other chronic diseases, where fluctuations in key parameters (such as blood pressure, serum glucose levels or rheumatic inflammation flares) can occur unpredictably at any time during treatment, lowering sUA enables us to cure gout without exception. Let’s finally make it happen!
CV: cardio-vascular
CVD: cardio-vascular disease
DECT: dual energy computerized tomography
MSU: mono-sodium urate
sUA: serum uric acid
ULD: uric acid lowering drug
ULT: uric acid lowering treatment
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
