
Research Article | DOI: https://doi.org/10.31579/2690-4861/541
College of Chemistry and Material Science, Shandong Agricultural University, Taian 271018, P. R. China.
*Corresponding Author: Nan Lu, College of Chemistry and Material Science, Shandong Agricultural University, Taian 271018, P. R. China.
Citation: Nan Lu and Chengxia Miao, (2024), Theoretical Investigation on Intramolecular Friedel-Crafts Alkylation Of 2,2′-Disubstituted 1,3-Indandione Delivering Axially Chiral Biaryls Catalyzed by Chiral Phosphoric Acid, International Journal of Clinical Case Reports and Reviews, 19(2); DOI:10.31579/2690-4861/541
Copyright: © 2024, Nan Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 16 September 2024 | Accepted: 30 September 2024 | Published: 07 October 2024
Keywords: central-to-axial conversion; Friedel-Crafts alkylation; rearrangement; aromatization; chiral phosphoric acid
Our Density Functional Theory (DFT) calculations provide the first theoretical investigation on CPA-catalyzed intramolecular Friedel-Crafts alkylation of 1,3-indandione. The reaction is initiated by bis-coordination of CPA with carbonyl and phenol of indandione. The promotion of CPA lies in H-bridge and steric effect from big silyl substituents confirming less-hindered Re-face more favorable than more-shielded Si-face within central chiral alcohol. Next AlCl3-promoted process contains four steps. The dehydration gives positive carbocation, the rearrangement of which activates another carbonyl. The water reversely splits into hydrogen and hydroxyl to recover phenol. The naphthol is obtained from opening of five-membered ring. The central-to-axial chirality conversion is secured between methyl/phenol substituents and carboxylic moiety of axially chiral biaryls. The positive solvation effect is suggested by decreased absolute and activation energies in solution compared with in gas. These results are supported by Multiwfn analysis on FMO composition of specific TSs, and MBO value of vital bonding, breaking.
The structural core of axially chiral bi(hetero)aryl are identified with potential medicinal applications in pharmaceuticals and as natural products [1–3]. This brings various strategies to handling stereochemistry from drug regulatory agencies. The ability of natural receptors possess differential binding between atropisomers and require new techniques for atropselective synthesis of desired targets. The general approach frequently used in asymmetric synthesis for chiral ligands and catalysts has received much attention such as phosphoramidites, the privileged ligands in asymmetric catalysis [4] and binaphthyl scaffold versatile in C–H functionalization [5]. Therefore the enantioselective synthetic method is attractive eliminating unwanted enantiomer and worthy significant efforts devoted to those chiral scaffolds. Bringmann reported atroposelective synthesis of axially chiral biaryl natural products. Kumarasamy analyzed nonbiaryl and heterobiaryl atropisomers as molecular templates [6]. Metrano achieved peptide-based catalysts through remote desymmetrization and atroposelectivity [7]. To synthesize enantio-enriched atropisomer, the central atropchiral framework is always controlled to construct stereogenic axis from non-chiral substrates using chiral catalyst [8-11]. Mei’s atropisomers beyond the C–C axial chirality, Li’s axially chiral indole-based frameworks and Zhang’s atropisomers bearing multiple chiral elements [12-14].Besides these methods, the central-to-axial chirality conversion including creation of stereogenic axis and simultaneous destruction of stereogenic center through aromatization become increasingly popular [15]. Thus it is desirable yet challenging to investigate the potential of carbocation generated in enantioselective desymmetrizing of 1,3-diones. Qin discovered desymmetric enantioselective reduction of cyclic 1,3-diketones catalyzed by recyclable phosphinamide organocatalyst [16]. Yang disclosed enantioselective condensation with hydrazine [17]. Barik reported NHC-catalyzed desymmetrization of N-aryl maleimides leading to atroposelective synthesis of N-aryl succinimides [18]. Ghosh obtained fischer indolization through dynamic kinetic resolution [19].The sophisticated carbocation is expected to be generated via dehydration, which undergoes energetically favored rearrangement and aromatization proposed by Brotschi’s oxadiazole derivatives as dual orexin receptor antagonists givingstructure–activity relationship [20]. In this field, the contribution of Liu group is remarkable in a recent breakthrough of chiral phosphoric acid (CPA)-catalyzed intramolecular Friedel-Crafts alkylation arising from 2,2′-disubstituted 1,3-indandione [21]. They also envisioned chiral alcohol featuring adjacent quaternary stereocenter would be generated catalyzed by Brönsted acid CPA. As we know, CPA was previously utilized in atroposelective synthesis of indole derivatives bearing axial chirality and enantioselective synthesis of 3-arylindole atropisomers via indolization of iminoquinones [22,23]. Although axially chiral biaryls were yielded in excellent yields, many problems still puzzled and there was no report about detailed mechanistic study explaining the central-to-axial chirality conversion. Since CPA was proposed to interact with phenol hydroxyl and carbonyl within substrate [24,25], What’s specific process of dual hydrogen-bonding activation modes? Why high stereoselectivity for central chiral alcohols was determined by the first desymmerization/functionalization of substrate? Was the construction of axial chirality influenced via Lewis acid (AlCl3)-promoted carbocation rearrangement? To solve these questions in experiment, an in-depth theoretical study was necessary for this strategy also focusing on the exploration of carbocation’s potential.
The geometry optimizations were performed at the B3LYP/BSI level with the Gaussian 09 package [26,27]. The mixed basis set of LanL2DZ for I and 6-31G(d) for other non-metal atoms [28-32] was denoted as BSI. Different singlet and multiplet states were clarified with B3LYP and ROB3LYP approaches including Becke's three-parameter hybrid functional combined with Lee−Yang−Parr correction for correlation [33,34]. The nature of each structure was verified by performing harmonic vibrational frequency calculations. Intrinsic reaction coordinate (IRC) calculations were examined to confirm the right connections among key transition-states and corresponding reactants and products. Harmonic frequency calculations were carried out at the B3LYP/BSI level to gain zero-point vibrational energy (ZPVE) and thermodynamic corrections at 353 K and 1 atm for each structure in toluene. The solvation-corrected free energies were obtained at the B3LYP/6-311++G(d,p) (LanL2DZ for I) level by using integral equation formalism polarizable continuum model (IEFPCM) in Truhlar’s “density” solvation model [35-37] on the B3LYP/BSI-optimized geometries. As an efficient method of obtaining bond and lone pair of a molecule from modern ab initio wave functions, NBO procedure was performed with Natural bond orbital (NBO3.1) to characterize electronic properties and bonding orbital interactions [38,39]. The wave function analysis was provided using Multiwfn_3.7_dev package [40] including research on frontier molecular orbital (FMO) and Mayer bond order (MBO).
The mechanism was explored for CPA-catalyzed intramolecular Friedel-Crafts alkylation of 2,2′-disubstituted 1,3-indandione 1 affording central chiral tertiary alcohol 2 and AlCl3-promoted carbocation rearrangement/aromatization delivering axially chiral biaryl-2-carboxylic acid 3 (Figure 1). Illustrated by Figure 2, the reaction is initiated by bis-coordination of CPA with carbonyl and phenol motif of 1 (black arrow). With bis-trimethyl silyl substituents of model CPA, the indandione 1 can be positioned in two ways allowing the subsequent intramolecular Friedel-Crafts alkylation to take place from less-hindered Re-face or more-shielded Si-face of carbonyl, leading to desymmetrization and stereoselectivity within tertiary alcohol 2. The “r” and “s” prefixes are used for stationary points on two parallel paths respectively. Two steps are located for this process that is nucleophilic attack and proton transfer. Then based on the coordination of phenol and hydroxyl of 2 with AlCl3 (red arrow), intermediate i3 is formed which undergoes dehydration assisted by AlCl3 giving positive C6 adjacent to quaternary chiral center C5 within carbocation i4. The rearrangement of i4 from the dissociation of AlCl3 with water affords i5, where another carbonyl residual is activated by AlCl3 (blue arrow). The water molecule reversely splits into hydrogen and hydroxyl group to recover phenol and link to positive C6 in i5-1. In next step the hydroxyl is bonded to C7 yielding carboxyl group of i5-2. Finally the conjugated naphthol structure is obtained from opening of five-membered ring with concerted double bond formation in axially chiral biaryls 3, in which the excellent central-to-axial chirality conversion is secured between methyl/phenol substituents and carboxylic moiety.
The schematic structures of optimized TSs in Figure 2 were listed by Figure 3. The activation energy was shown in Table 1 for all steps. Supplementary Table S1, Table S2 provided the relative energies of all stationary points. According to experiment, the Gibbs free energies in toluene solution phase are discussed here [21]. However, in order to save computing resources, methyl was used to replace phenyl group of catalyst CPA in experiment. This makes an absolutely accurate prediction of stereoselectivities unavailable for this study.
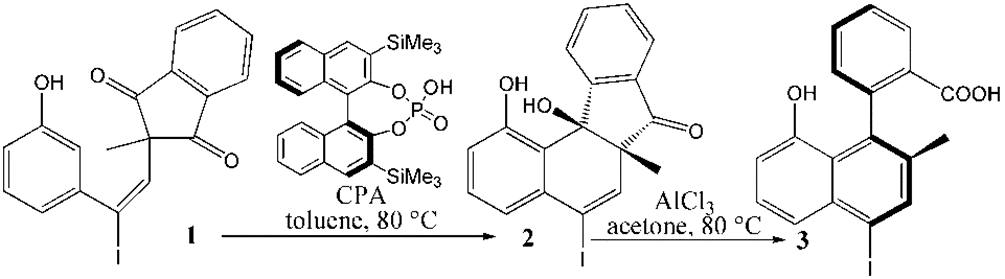
Figure 1: CPA-catalyzed intramolecular Friedel-Crafts alkylation of 2,2′-disubstituted 1,3-indandione 1 affording central chiral tertiary alcohol 2 and AlCl3-promoted carbocation rearrangement/aromatization delivering axially chiral biaryl-2-carboxylic acid 3.
| Species | ΔGgas | ΔGsol(toluene) |
| 1 | 0.00 | 0.00 |
| 2 | -5.60 | -3.78 |
| 3 | -25.79 | -23.86 |
| 1+CPA | 0.00 | 0.00 |
| r-i1 | -412.88 | -412.13 |
| r-tsi12-0 | -388.65 | -387.88 |
| r-i1-1 | -396.98 | -395.01 |
| r-tsi12-1 | -360.86 | -359.52 |
| r-i2 | -413.67 | -419.65 |
| s-i1 | -414.89 | -413.46 |
| s-tsi12-0 | -384.26 | -381.86 |
| s-i1-1 | -396.78 | -395.20 |
| s-tsi12-1 | -360.88 | -359.63 |
| s-i2 | -406.68 | -408.69 |
| 1+AlCl3 | 0.00 | 0.00 |
| i3 | -101.87 | -101.47 |
| tsi34 | -94.97 | -96.59 |
| i4 | -106.58 | -108.16 |
| i5 | -98.09 | -99.90 |
| tsi56-0 | -80.42 | -82.43 |
| i5-1 | -111.56 | -112.99 |
| tsi56-1 | -82.98 | -85.11 |
| i5-2 | -87.95 | -93.40 |
| tsi56-2 | -86.29 | -90.31 |
| i6 | -134.65 | -134.48 |
Table S1. Calculated relative energies (all in kcal mol-1, relative to isolated species) for the ZPE-corrected Gibbs free energies (ΔGgas), Gibbs free energies for all species in solution phase (ΔGsol) at 353 K by B3LYP/6-311++G(d,p)//B3LYP/6-31G(d) method and difference between absolute energy.
| TS | ΔG≠gas | ΔG≠sol |
| r-tsi12-0 (294i) | 24.2 | 24.2 |
| r-tsi12-1 (1393i) | 36.1 | 35.5 |
| s-tsi12-0 (282i) | 30.6 | 31.6 |
| s-tsi12-1 (1398i) | 35.9 | 35.6 |
| tsi34 (167i) | 6.9 | 4.9 |
| tsi56-0 (508i) | 17.7 | 17.4 |
| tsi56-1 (390i) | 28.6 | 27.9 |
| tsi56-2 (234i) | 1.7 | 3.1 |
Table S2. The activation energy (local barrier) (in kcal mol−1) of all reactions in the gas, solution phase calculated with B3LYP/6-311++G(d,p)//B3LYP/6-31G(d) method.
| C1···C6 | O3···H1 | O2···H2 | ||
r-tsi12-0 | 0.52 | 0.23 | 0.46 | |
| C1···H3 | H3···O2 | O2···H2 | H2···O4 | |
| r-tsi12-1 | 0.50 | 0.29 | 0.23 | 0.49 |
| O1···H1 | H1···O2 | C6···O2 | ||
| ts-i34 | 0.46 | 0.39 | 0.25 | |
| O1···H1 | H1···O2 | C6···O2 | ||
| tsi56-0 | 0.27 | 0.49 | 0.39 | |
| C6···O2 | O2···C7 | |||
| tsi56-1 | 0.37 | 0.54 | ||
| C5···C7 | C5···C6 | |||
| tsi56-2 | 0.62 | 1.25 |
Table S3. Mayer bond order (MBO) of typical TSs
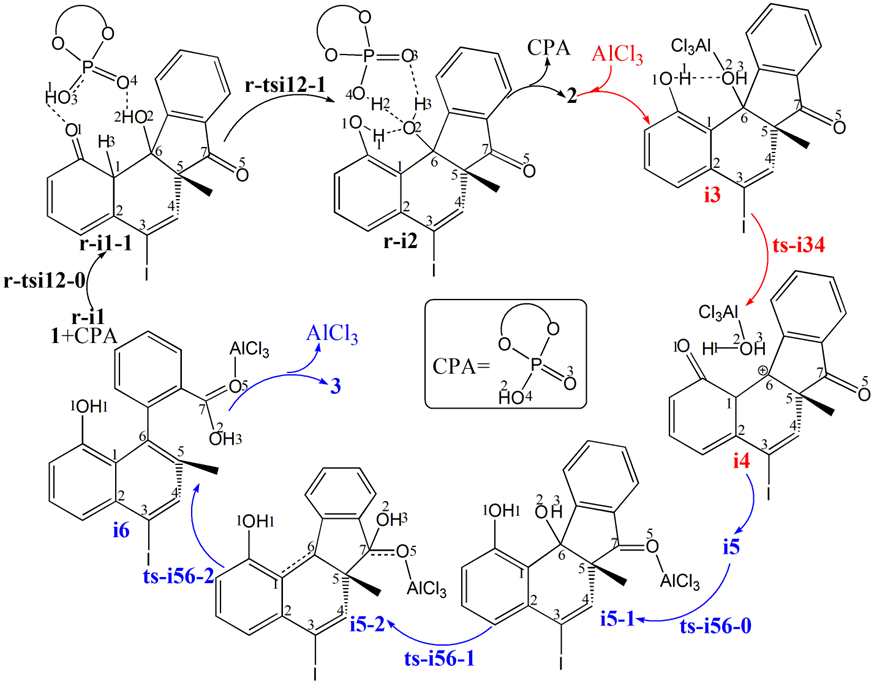
Figure 2: Proposed reaction mechanism of CPA-catalyzed intramolecular Friedel-Crafts alkylation of 1 affording 2 and AlCl3-promoted carbocation rearrangement/aromatization delivering 3. TS is named according to the two intermediates it connects.
| TS | ΔG≠gas | ΔG≠sol |
| r-tsi12-0 | 24.2 | 24.2 |
| r-tsi12-1 | 36.1 | 35.5 |
| s-tsi12-0 | 30.6 | 31.6 |
| s-tsi12-1 | 35.9 | 35.6 |
| tsi34 | 6.9 | 4.9 |
| tsi56-0 | 17.7 | 17.4 |
| tsi56-1 | 28.6 | 27.9 |
| tsi56-2 | 1.7 | 3.1 |
Table 1: The activation energy (in kcal mol−1) of all reactions in gas and solvent
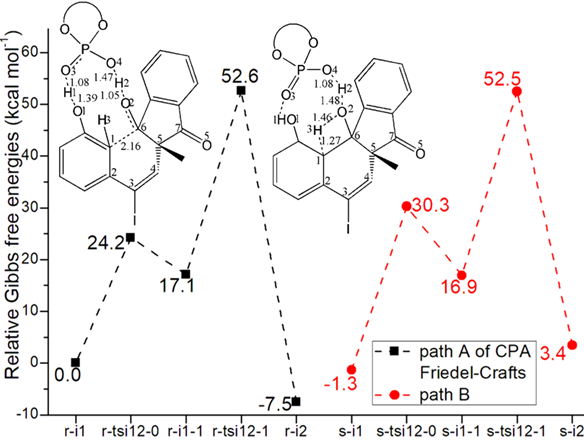
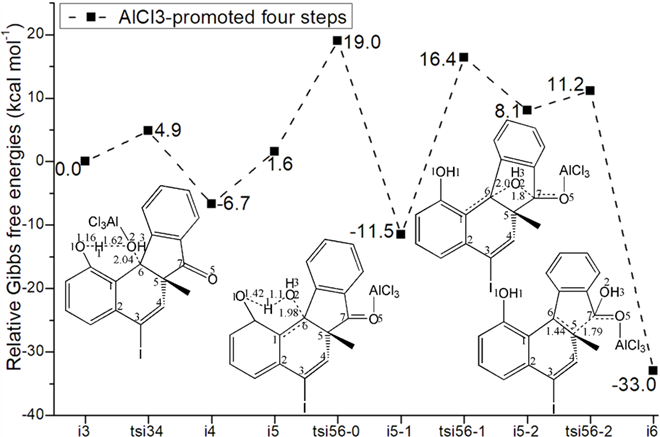
Figure 3: Relative Gibbs free energy profile in solvent phase starting from complex (a) r-i1 (b) i3 (Bond lengths of optimized TSs in Å).
3.1 CPA-catalyzed intramolecular Friedel-Crafts alkylation
Two steps are located for the first phaseof CPA-catalyzed intramolecular Friedel-Crafts alkylation. Owing to the same process, “r” stationary points from less-hindered Re-face will be discussed in detail denoted as path A (black dash line of Figure 3a). The initial complex r-i1 is stabilized by bis-coordination of CPA with carbonyl and phenol motif of 1 forming H bond depending on P-O4H2 and P=O3. The intramolecular nucleophilic addition proceeds via r-tsi12-0 in step 1 with the activation energy of 24.2 kcal mol−1 relative to the starting point r-i1 endothermic by 17.1 kcal mol−1 producing r-i1-1. The transition vector containstwo parts in concerted modes that is dual proton transfer of H1 from O1 to O3 and H2 from O4 to O2 followed by approaching of negative C1 to positive C6 (1.39, 1.08, 1.47, 1.05, 2.16 Å) (Figure S1a). Obviously with formal C1-C6 single bond, the new six-membered ring in r-i1-1 is reactive to initiate next step.
Then mediated by CPA, the proton transfer occurs via r-tsi12-1 as step 2 with activation energy of 35.5 kcal mol−1 exothermic by -7.5 kcal mol−1 giving intermediate r-i2. The transition vector includes proton H3 on sp3 hybrid C1 moving to O2 along with cooperated returing of H2 from O2 to O4 (1.27, 1.46, 1.48, 1.08 Å) (Figure S1b). In stable r-i2, O2H3 not only forms bis-coordination with CPA via two H bonds but makes inter O1-H1···O2 available. In addition, C1 recovers sp2 hybrid of phenol after kicking off H3. Clearly during two steps of thefirst phase, CPA constantly functions as LA providing H-bridge driving force. When Friedel-Crafts alkylation is completed, the central chiral tertiary alcohol 2 is obtained after removal of CPA.
3.2 AlCl3-promoted dehydration/carbocation rearrangement/carboxyl formation/aromatization
Four steps are located in next phase promoted by AlCl3, which plays role of BA acceptinglone pair of hydroxyl or carbonyl. The initial intermediate i3 is formed with Al-O2H3 coordination taken as new starting point of the four steps (black dash line of Figure 3b). Assisted by AlCl3, the dehydration undergoes in step 3 via ts-i34 with activation energy of 4.9 kcal mol−1 affording i4 exothermic by -6.7 kcal mol−1. According to the transition vector, O2H3 and H1 donated by phenol O1 assembles water molecule, which is ruptured from C6 afterwards that is remarkable O1···H1···O2 and C6···O2 cleavage (1.16, 1.62, 2.04 Å) (Figure S1c). Taken away of water by AlCl3 affords positive C6 adjacent to quaternary chiral center C5 within carbocation i4. Subsequently, the rearrangement of i4 generates reactive i5 with increased relative energy. This is achieved through dissociation of AlCl3 with water andturns to activate another carbonyl residual C7=O5 forming Al-O5 single bond. In step 4, the water molecule H1O2H3 reversely splits into hydrogen H1 and hydroxyl O2H3 to recover phenol and link to positive C6. An intermediate i5-1 is delivered via tsi56-0 with activation energy of 17.4 kcal mol−1 exothermic by -11.5 kcal mol−1 . The transition vector corresponds to O2···H1···O1 and O2···C6 linkage (1.1, 1.42, 1.98 Å) just opposite to the case of ts-i34 (Figure S1d). Although characterized by similar structure with i3, i5-1 becomes more stable with lower energy indicating this worthy transformation. Next, hydroxyl O2H3 moves from C6 to C7 via tsi56-1 instep 5 with activation energy of 27.9 kcal mol−1 endothermic by 8.1 kcal mol−1 yielding carboxyl group of i5-2. The transition vectorreveals breaking of O2···C6 and bonding of O2-C7 (2.0, 1.8 Å) (Figure S1e). i5-2 is quite reactive ready for the final step. Consequently, the opening of five-membered ring proceeds via tsi56-2 in step 6 with small activation energy of 3.1 kcal mol−1 enormously exothermic by -33.0 kcal mol−1. In resultant i6, the conjugated naphthol structure is obtainedvia aromatization that is simultaneous new C5-C6 double bond formation. The detailed atomic motion is illustrated according to the transition vector about dissociation of C5-C7 and shortening of C5-C6 bond from single to double (1.79, 1.44 Å) (Figure S1f). Once AlCl3 is left, axially chiral biaryls 3 is achieved denoting central-to-axial chirality conversion between methyl/phenol substituents and carboxylic moiety. Comparatively, proton transfer mediated by CPA of step 2 is determined to be rate-limiting for the whole process. To highlight the idea of feasibility for changes in electron density and not molecular orbital interactions are responsible of the reactivity of organic molecules, quantum chemical tool Multiwfn was applied to analyze of electron density such as MBO results of bonding atoms and contribution of atomic orbital to HOMO of typical TSs (Table S3, Figure S2). These results all confirm the above analysis.
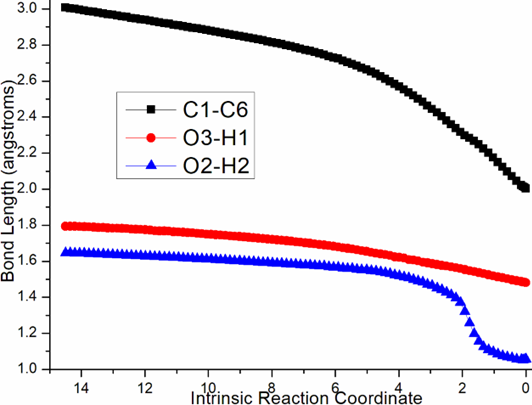
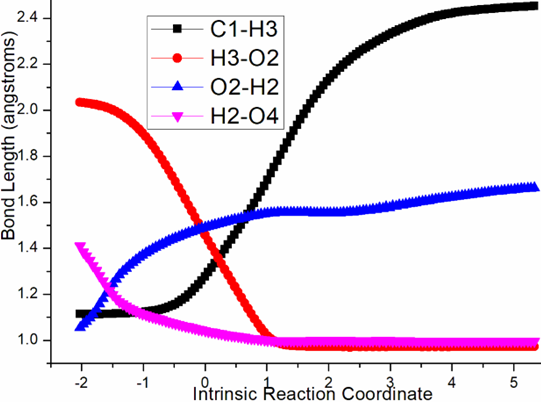
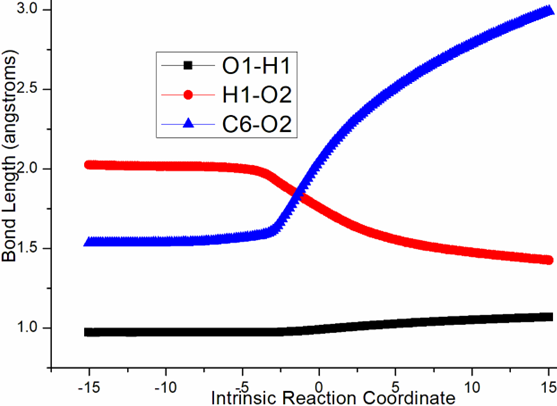
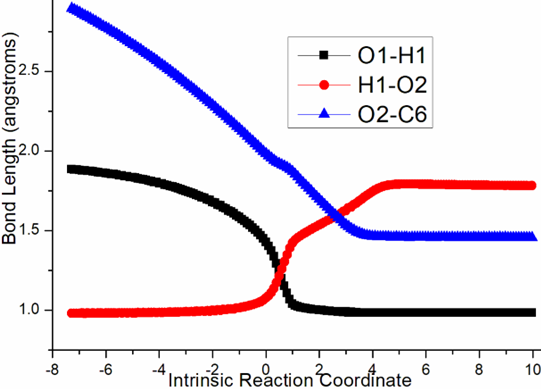
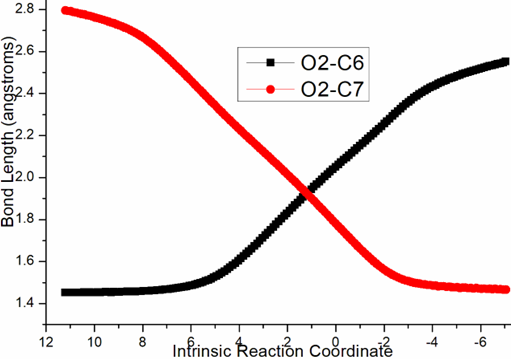
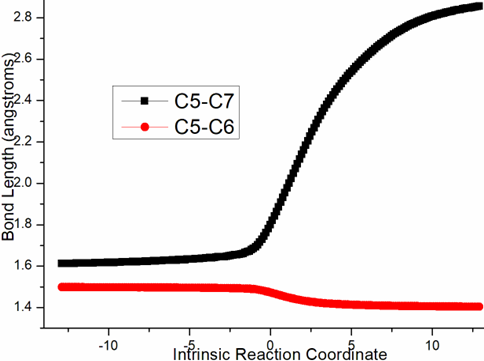
Figure S1. Evolution of bond lengths along the IRC for (a) r-tsi12-0 (b) r-tsi12-1 (c) ts-i34 (d) tsi56-0 (e) tsi56-1 (f) tsi56-2 at B3LYP/6-311++G(d,p) level.
Our DFT calculations provide the first theoretical investigation on CPA-catalyzed intramolecular Friedel-Crafts alkylation of 2,2′-disubstituted 1,3-indandione. The reaction is initiated by bis-coordination of CPA with carbonyl and phenol of indandione via two steps of nucleophilic attack and proton transfer with the latter determined to be rate-limiting. The promotion of CPA not only lies in H-bridge driving force but steric effect attributed by big silyl substituents confirming less-hindered Re-face more favorable than more-shielded Si-face both from kinetics and thermodynamics leading to desymmetrization and stereoselectivity within central chiral alcohol. Next as BA accepting lone pair of hydroxyl or carbonyl, AlCl3-promoted process contains four steps. The dehydration undergoes giving positive carbocation. The rearrangement of carbocation arises from dissociation of water with AlCl3, which activates another carbonyl. The water molecule reversely splits into hydrogen and hydroxyl to recover phenol. Then hydroxyl turns from positive carbon to carbonyl carbon yielding carboxyl group. Finally the conjugated naphthol is obtained from opening of five-membered ring with concerted double bond formation. The central-to-axial chirality conversion is secured between methyl/phenol substituents and carboxylic moiety of axially chiral biaryls. The positive solvation effect is suggested by decreased absolute and activation energies in toluene solution compared with in gas. These results are supported by Multiwfn analysis on FMO composition of specific TSs, and MBO value of vital bonding, breaking.
Electronic Supplementary Material
Supplementary data available: [Computation information and cartesian coordinates of stationary points; Calculated relative energies for the ZPE-corrected Gibbs free energies (ΔGgas), and Gibbs free energies (ΔGsol) for all species in solution phase at 353 K.]
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
