
Research Article | DOI: https://doi.org/10.31579/2690-1897/202
College of Chemistry and Material Science, Shandong Agricultural University, Taian City 271018, Shandong Prov., P.R. China.
*Corresponding Author: Nan Lu, Shandong Agricultural University, Taian City 271018, Shandong Prov., P.R. China.
Citation: Nan Lu, Chengxia Miao, (2024), Theoretical investigation on Brønsted acid-catalyzed cascade ring open-ing and double cyclization of 3-ethoxy cyclobutanone with naphthol to synthesize 2,8-dioxabicyclo [3.3.1] nonane, J, Surgical Case Reports and Images, 7(6); DOI:10.31579/2690-1897/202
Copyright: © 2024, Nan Lu. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 01 July 2024 | Accepted: 12 July 2024 | Published: 18 July 2024
Keywords: Naphthol; ring-opening; cyclobutanone; (3+3) annulation; bicyclic ketal
The mechanism is investigated for cascade ring-opening/cyclization of 3-ethoxy 2-phenyl cyclobutanone with 2-naphthol leading to 2,8-dioxabicyclo[3.3.1]nonane. After protonation of carbonyl group, the nucleophilic attack of 2-naphthol prompts ring-opening of cyclobutanone. The enolization and ethanol removal generate alkenone with recovered naphthalene hydroxyl, which follows two possible pathways. In path A, another 2-naphthol facilitates Michael addition to render enol intermediate, which then affords desired product via dioxygen double cyclization containing three steps. In path B, the protonation from hydroxyl helps cyclization furnishing dihydropyrylium. With another deprotonated 2-naphthol, (3+3) annulation directly forms product and is more favorable than path A. Under the impact of Brønsted acid, positive (3+3) annulation is further improved with low barrier. The positive solvation effect is suggested by decreased absolute and activation energies in solution compared with in gas. These results are supported by Multiwfn analysis on FMO composition of specific TSs, and MBO value of vital bonding, breaking.
The 2,8-dioxabicyclo[3.3.1]nonanes are privileged members in numerous heterocyclic frameworks embedded with two oxygen atoms [1,2]. Since these bicyclic moieties constitute flavonoid compounds, they exhibit powerful anti-inflammatory, antiviral, antioxidant, and anticancer activities as several important drugs [3,4]. Characterized by rigid V-shaped diaryl-substituted structure, the construction of these bicyclic cores has received wide interest in recent years [5]. In this aspect, most methods and protocols require similar precursor of 2-hydroxy chalcones with different partners such as sequential knoevenagel condensation and hetero-Diels-Alder reaction in aqueous medium, cerium(III)-catalyzed domino approach using polyphenols and 1,4-Zwitterionic intermediates generated by regioselective cleavage of cyclobutane ring followed by their cycloaddition [6-8]. Then Zhu reported cationic-lanthanide-complex-catalyzed reaction with naphthols [9]. Wang developed controllable synthesis of two isomers 4H chromene under catalyst-free conditions [10]. Li achieved switchable synthesis of cyclohexanedione-fused 2,8-oxaza/2,8-dioxa bicyclo[3.3.1]nonanes by tunability of deamination/dehydration process [11].
The 1,4-zwitterionic intermediate generated from cleavage of C2−C3 bond was common before various ring opening, cycloaddition or rearrangement catalyzed by Lewis acid. In past few decades, many examples appeared such as intermolecular [4 + 2] cycloaddition of 3-alkoxycyclobutanone to aldehyde and ketone, synthesis of pyrazole through union of monosubstituted hydrazine with 3-ethoxycyclobutanone, total synthesis of (±)-aspidospermidine and regioselective inter-and intramolecular formal [4 + 2] cycloaddition of cyclo-butanone with indole, [3 + 3] annulation to construct pyrimidine and pyridine, between aromatic amine and 3-ethoxycyclobutanone to 2-lkylquinoline, remote site-selective Friedel-Crafts alkylation of β-Naphthol [12-17]. Okado achieved (±)-bremazocine, 2,3-di- or 2,3,3-trisubstituted N-Ts-2,3-dihydro-4-pyridone by using reaction of N-p-Toluenesulfonyl (Ts) aldimine with 3-ethoxycyclobutanone prompted by titanium(IV) chloride [18]. Lin reported diversity-oriented synthesis of bioactive pyridine-containing fused heterocycle through union of 3-ethoxycyclobutanone with various heterocyclic amine with excellent regioselectivity [19]. In despite of these progresses, activation of four-membered ring via Brønsted acid (BA) is few. Only Xu obtained enantioselective Baeyer–Villiger oxidation of 3-substituted cyclobutanone giving γ-lactone [20].
However, it is still rare for direct ring-opening or cyclization with 3-ethoxy cyclobutanone catalyzed by BA. As far as we know, the development in recent years was Rahmatpour’s regioselective synthesis of di-aromatic ring-fused 2,8-dioxa/dithia bicyclo[3,3,1]nonane via recyclable tandem formation of multiple chemical C-C/C-O and C-C/C-S bonds [21]. In this context, a breakthrough was Hazra’s cascade ring-opening/cyclization of 3-ethoxy cyclobutanone [22]. Furthermore, this type of complex 15-membered macrocycle containing heteroatom was important biologically showing inhibitory and antitumor activities [23-25]. Although a range of 2,8-dioxabicyclo[3.3.1]nonane were obtained with various substituted naphthol, many problems still puzzled and there was no report about detailed mechanistic study explaining BA activation. What is the priority for carbonyl protonation of cyclobutanone and polarization of C2−C3 bond? How double cyclization is initiated from two oxygen atoms of two naphthols after Michael addition? Why (3+3) annulation with another 2-naphthol also possible from dihydropyrylium-type intermediate after cyclization from hydroxyl group? To solve these questions in experiment, an in-depth theoretical study was necessary for this strategy focusing on the competition of dioxygen-initiated double cyclization and (3+3) annulation.
Optimized structures were obtained at M06-2X/6-31G(d) level of theory with GAUSSIAN09 [26]. In tests of popular DFT methods [27], M06-2X functional attained smaller standard deviation of difference between calculated value and experimental value in geometries than B3LYP including Becke's three-parameter hybrid functional combined with Lee−Yang−Parr correction for correlation [28,29]. The best compromise between accuracy and time consumption was provided with 6-31G(d) basis set on energy calculations. Also, M06-2X functional was found to give relatively accurate results for catalysed enantioselective (4 + 3), concerted [4 + 2], stepwise (2 + 2) cycloaddition and catalysed Diels−Alder reactions [30,31]. Together with the best performance on noncovalent interaction, M06-2X functional is believed to be suitable for this system [32-34]. The nature of each structure was verified by performing harmonic vibrational frequency calculations. Intrinsic reaction coordinate (IRC) calculations were examined to confirm the right connections among key transition-states and corresponding reactants and products. Harmonic frequency calculations were carried out at the M06-2X/6-31G(d) level to gain zero-point vibrational energy (ZPVE) and thermodynamic corrections at 298.15 K and 1 atm for each structure in toluene.
The solvation-corrected free energies were obtained at the M06-2X/6-311++G(d,p) level by using integral equation formalism polarizable continuum model (IEFPCM) in Truhlar’s “density” solvation model [35-39] on the M06-2X/6-31G(d)-optimized geometries. As an efficient method obtaining bond and lone pair of a molecule from modern ab initio wave functions, NBO procedure was performed with Natural bond orbital (NBO3.1) to characterize electronic properties and bonding orbital interactions [40-42]. The wave function analysis was provided using Multiwfn_3.7_dev package [43] including research on frontier molecular orbital (FMO) and Mayer bond order (MBO).
The mechanism was explored for BA-catalyzed cascade ring-opening/cyclization of 3-ethoxy 2-phenyl cyclobutanone 1 with 2-naphthol 2 leading to 2,8-dioxabicyclo [3.3.1] nonane 3 (Scheme 1). Illustrated by black arrow of Scheme 2, initially the carbonyl group protonation of 1 polarize the C2−C5 bond. Then via the nucleophilic attack of 2 at C2 center, the ring-opening of 1 happen leading to intermediate I. Eventually the enolization and removal of ethanol generate intermediate II, which follows two possible pathways. In path A, another molecule 2 facilitates Michael addition to render intermediate III, which then affords desired product 3 via double cyclization from two oxygen atoms of two molecules of 2 (red arrow). In path B, the protonation of II carbonyl group by hydroxyl of 2 facilitates cyclization furnishing dihydropyrylium. Subsequently, a (3+3) annulation with another deprotonated 2 leads to product 3 (blue arrow). Under the impact of BA the positive (3+3) annulation takes place via three steps (green arrow). The schematic structures of optimized TSs in Scheme 2 were listed by Figure 1. The activation energy was shown in Table 1 for all steps. Supplementary Table S1, Table S2 provided the relative energies of all stationary points. According to experiment, the Gibbs free energies in methanol solution phase are discussed here.
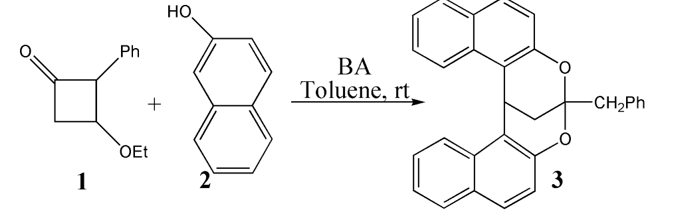
Scheme 1 (a) Brønsted acid (BA)-catalyzed cascade ring-opening/cyclization of 3-ethoxy 2-phenyl cyclobutanone 1 with 2-naphthol 2 leading to 2,8-dioxabicyclo [3.3.1] nonane 3.
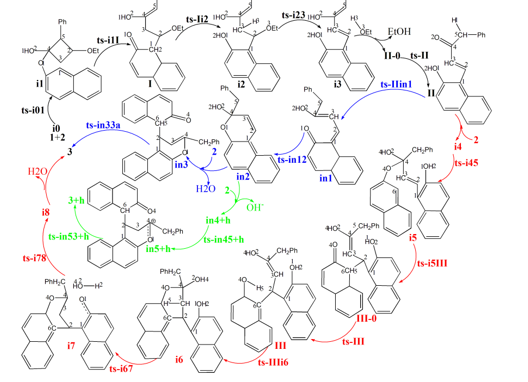
Scheme 2: Proposed reaction mechanism of BA-catalyzed cascade ring-opening/cyclization of 1 with 2 leading to 3. TS is named according to the two intermediates it connects.
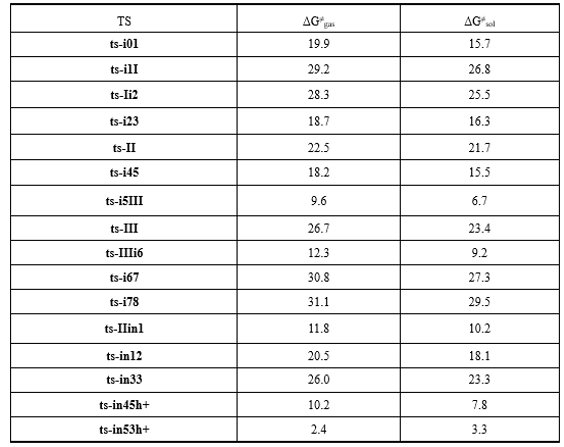
Table 1: The activation energy (in kcal mol−1) of all reactions in gas and solvent
(b)
(c)
3.1 Protonation/ring-opening/enolization/ethanol removal and enol isomerization
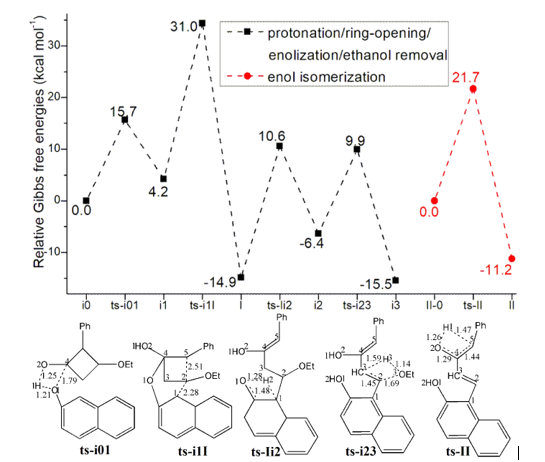
Initially the complex of 1 and 2 denoted as i0, the carbonyl group of 1 is protonated by hydroxyl of 2 polarizing the C2−C5 bond. This process proceeds via ts-i01 as step 1 with the activation energy of 15.7 kcal mol−1 relative to the starting point i0 endothermic by 4.2 kcal mol−1 (black dash line of Figure 1a). The transition vector includes the transfer of H1 from O1 to O2 and the closing of O1 to C4 makes it changing from sp2 hybrid to sp3 (1.21, 1.25, 1.79 Å) (Figure S1a). Without proton, the reactive hydroxyl O1 is bonded to sp3 C4 in resultant i1, from which the ring-opening of 1 happen under the nucleophilic attack of 2 at C2 center leading to intermediate I. As step 2, this occurs via ts-i1I with activation energy of 26.8 kcal mol−1 exothermic by -14.9 kcal mol−1. The transition vector contains cleavage of C2···C5 and concerted linkage of C1···C2 (2.28, 2.51 Å). Therefore I is stable after releasing the tension of quadruple ring of 1. While once bonded to C2, the C1 turns to be sp3 hybrid breaking the conjugation of naphthalene ring apt to kick off a hydrogen in following process.
Then the enolization of carbonyl O1 takes place via ts-Ii2 in subsequent step 3 with activation energy of 25.5 kcal mol−1 affording i2 exothermic by -6.4 kcal mol−1. According to the transition vector, the proton H2 is transferring from C1 to O1, which is recovered to hydroxyl group O1H2. In next step 4, the OEt bonded to C2 and H on C3 are disconnected simultaneously and assemble stable EtOH molecule leaving from the system. Via ts-i23, ethanol is removed with a barrier of 16.3 kcal mol−1 exothermic by -15.5 kcal mol−1 delivering intermediate i3. The transition vector corresponds to the elongation of C2-O3, C3-H3 single bond to dissociation and bonding of O3-H3, C2-C3 shortened from single to double one at the same time (1.69, 1.59, 1.14, 1.45 Å) (Figure S1b).
Without ethanol, the intermediate II-0 is taken as new starting point of step 5 (red dash line of Figure 1a). The O2H1 hydroxyl group formed in step 1 denotes H1 to sp2 C5 to complete enol tautomerism. This happens via ts-II with activation energy of 21.7 kcal mol−1 exothermic by -11.2
kcal mol−1 yielding intermediate II more stable. The transition vector suggests remarkable O2···H1···C5 shifting as well as C4-C5 stretching from double bond to single.
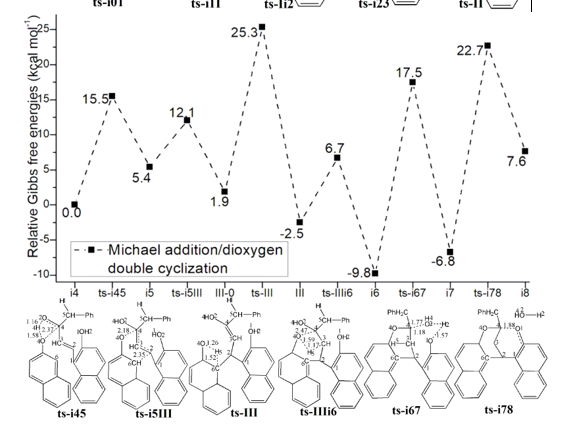
3.2 Michael addition/dioxygen double cyclization
Two pathways are possible from II. In path A (black dash line of Figure 1b), the addition of another molecule 2 forms complex i4 as starting point of the following six steps. As preparation of Michael addition, the step 6 takes place via ts-i45 with activation energy of 15.5 kcal mol−1 forming reactive i5 endothermic by 5.4 kcal mol−1. Similar with ts-i01, the carbonyl O2 is protonated by new naphthyl hydroxyl O4H4. According to the transition vector, the atomic motion is composed of O4···H4···O2 proton shifting and laggered approaching of O4···C4 (1.58, 1.16, 2.37 Å). This greatly facilitate the crucial Michael addition seen from the low barrier via hexagonal ring structure of ts-i5III. Seen from the detailed motion demonstrated by its transition vector (Figure S1c), the departure of O4-C4 bond is accompanied by formation of C6-C2 bond (2.18, 2.35 Å). Clearly, the cleavage of O4-C4 endows negative charge concentrated on O4, passes on to C6 and enhance its nucleophilic ability. Likewise, the ability of C2 as an electrophilic center is also increased. Hence the small activation energy of step 7 is only 6.7 kcal mol−1 endothermic by 1.9 kcal mol−1 giving III-0, which is lower than former i5 in relative energy yet still reactive to initiate next step.
In step 8, III-0 undergoes carbonyl enolization of the second 2 via ts-III just like ts-Ii2 with activation energy of 23.4 kcal mol−1 exothermic by -2.5 kcal mol−1 generating III favorable in thermodynamics. The transition vector of this second enolization is about proton transfer of C6···H5···O4 (1.52, 1.26 Å), which not only recovers hydroxyl O4H5 but turns sp3 C6 in III-0 to be sp2 stable conjugated structure of naphthalene ring in III. From III, the desired product 3 is produced via double cyclization from two oxygen of two 2 molecule through three steps. The first nucleophilic attack of O4 to C4 proceeds via ts-IIIi6 followed by proton H5 given by O4 to C3 concertedly. This can be illustrated in detail by the transition vector (1.59, 1.17, 2.47 Å), which makes C3 methylene on the one hand and improves the nucleophilic ability of O4 on the other. The activation energy of step 9 is 9.2 kcal mol−1exothermic by -9.8 kcal mol−1 relative to III.
Before the second nucleophilic addition of O1 to C4, the dehydration is located via ts-i67 with activation energy of 27.3 kcal mol−1 exothermic by -6.8 kcal mol−1 as step 10. This is also belongs to small molecule H2O assembling and then leaving process similar to the case of EtOH removal via ts-i23. Here, the O2H4 on C4 combines with H2 on O1 as one water (1.77, 1.57, 1.18 Å) (Figure S1d) in resulting i7, which is stable although O1 and C4 is not yet connected owing to the distance. The real O1-C4 single bond should be achieved in last step 11 via ts-i78 with activation energy of 29.5 kcal mol−1 endothermic by 7.6 kcal mol−1 only involving simple O1···C4 approaching (1.88 Å). The barrier is somewhat high due to the need of overcoming tension generated by spatial distance.
Ultimately, the third simple step of dioxygen double cyclization is determined to be rate-limiting for path A. To highlight the idea of feasibility for changes in electron density and not molecular orbital interactions are responsible of the reactivity of organic molecules, quantum chemical tool Multiwfn was applied to analyze of electron density such as MBO results of bonding atoms and contribution of atomic orbital to HOMO of typical TSs (Table S3, Figure S2). These results all confirm the above analysis.
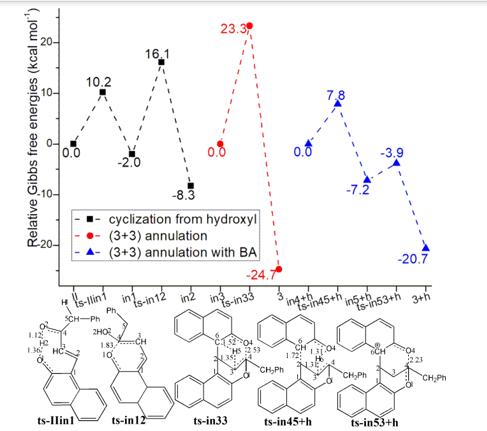
3.3 Cyclization from hydroxyl/(3+3) annulation and BA impact
In path B, the carbonyl O2 can also be protonated by original hydroxyl O1H2 from II via ts-IIin1 in step 6 with a barrier of 10.2 kcal mol−1 exothermic by -2.0 kcal mol−1delivering in1. The transition vector corresponds to simple proton H2 transfer from O1 to O2 (1.36, 1.12 Å) (Figure S1e). This faciliates first cyclization from O1 to C4 via ts-in12 as step 7 with the activation energy of 18.1 kcal mol−1 relative to in1 exothermic by -8.3 kcal mol−1 (black dash line of Figure 1c). Therefore the difficult nucleophilic addition of O1 to C4 in path A becomes readily accessible in path B, which is approved by the transition vector of ts-in12.
After the removal of O2H2 from cyclization product in2, a dihydropyrylium is obtained, which is easily binding the second deprotonated 2 via C6-C2 bond forming in3 as new starting point of subsequent step 3 (red dash line of Figure 1c). The second cyclization from O4 to C4 occurs via ts-in33 with activation energy of 23.3 kcal mol−1 by exothermic -24.7 kcal mol−1 directly leading to 3. The transition vector reveals a concerted asynchronous process containing previous C3 methylenation by C6 via C6···H5···C3 atomic motion and O4···C4 linking afterwords (1.52, 1.35, 2.53 Å) (Figure S1f). Evidently for path B, (3+3) annulation includes two steps with the second concerted one as rate-limiting more favorable than that of path A from kinetics.
The impact of BA are also considered for preferential (3+3) annulation modeled as a system with one positive charge (blue dash line of Figure 1c). The initial complex binding dihydropyrylium and neutral 2 denoted as in4+h is taken as starting point of the next two steps. The subsequent step 3 takes place via ts-in45+h with activation energy of 7.8 kcal mol−1 affording in5+h exothermic by -7.2 kcal mol−1, where C3 is methylenated by O4 via O4···H6···C3 and C6···C2 is bonding in concerted mode. Then via ts-in53+h with a rather low barrier of 3.3 kcal mol−1, O4-C4 single bond is accomplished in step 4 delivering 3+h exothermic by -20.7 kcal mol−1 as precusor of 3. Assisted by BA, the (3+3) annulation becomes easier involving three steps with the first O1···C4 bonding as rate-limiting comparatively.
Our DFT calculations provide the first theoretical investigation on cascade ring-opening/cyclization of 3-ethoxy 2-phenyl cyclobutanone with 2-naphthol leading to 2,8-dioxabicyclo[3.3.1]nonane. The carbonyl group of cyclobutanone is initially protonated by hydroxyl of 2-naphthol, the nucleophilic attack of which prompts ring-opening of cyclobutanone. The subsequent enolization and removal of ethanol generate alkenone intermediate with recovered naphthalene hydroxyl, which follows two possible pathways. In path A, another 2-naphthol facilitates Michael addition to render enol intermediate, which then affords desired product via dioxygen double cyclization containing three steps with the third simple O1···C4 approaching as rate-limiting. The barrier is somewhat high due to the tension generated by spatial distance yet readily to overcome under room temperature. In path B, the protonation from hydroxyl helps cyclization furnishing dihydropyrylium. With another deprotonated 2-naphthol, (3+3) annulation includes two steps with the second concerted O4···C4 bonding and C3 methylenation by C6 as rate-limiting, which directly forms product and is more favorable than path A from kinetics. Under the impact of BA the positive (3+3) annulation involves three steps with the first O1···C4 bonding as rate-limiting comparatively. The positive solvation effect is suggested by decreased absolute and activation energies in toluene solution compared with in gas. These results are supported by Multiwfn analysis on FMO composition of specific TSs, and MBO value of vital bonding, breaking.
Supplementary data available: [Computation information and cartesian coordinates of stationary points; Calculated relative energies for the ZPE-corrected Gibbs free energies (ΔGgas), and Gibbs free energies (ΔGsol) for all species in solution phase at 298 K.]
Conceptualization, Nan Lu; Methodology, Nan Lu; Software, Nan Lu; Validation, Nan Lu; Formal Analysis, Nan Lu; Investigation, Nan Lu; Resources, Nan Lu; Data Curation, Nan Lu; Writing-Original Draft Preparation, Nan Lu; Writing-Review & Editing, Nan Lu; Visualization, Nan Lu; Supervision, Chengxia Miao; Project Administration, Chengxia Miao; Funding Acquisition, Chengxia Miao. All authors have read and agreed to the published version of the manuscript.
This work was supported by National Natural Science Foundation of China (21972079) and Key Laboratory of Agricultural Film Application of Ministry of Agriculture and Rural Affairs, P.R. China.
The authors declare no conflict of interest.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
