
Research Article | DOI: https://doi.org/10.31579/2690-1897/217
College of Chemistry and Material Science, Shandong Agricultural University, Taian 271018, P. R. China
*Corresponding Author: Nan Lu, College of Chemistry and Material Science, Shandong Agricultural University, Taian 271018, P. R. China
Citation: Nan Lu, Chengxia Miao, (2024), Theoretical investigation on BF3·OEt2-catalyzed tandem benzannula-tion/Friedel−Crafts reaction of 2-alkynylaniline and 2-alkynylbenzaldehyde delivering highly π‑extended dihydroben-zo[a]indenocarbazole, J, Surgical Case Reports and Images, 7(9); DOI:10.31579/2690-1897/217
Copyright: © 2024, Nan Lu. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 24 September 2024 | Accepted: 10 October 2024 | Published: 16 October 2024
Keywords: Lewis Acid; benzannulation; friedel−crafts; π-extended; carbazole
Our DFT calculations provide the first theoretical investigation on BF3·OEt2-facilitated benzannulation/Friedel−Crafts reaction of 2-(phenylethynyl) aniline with 2-(phenylethynyl)benzaldehyde. Imine is formed mediated by BF3·OEt2 via initial two steps. The subsequent nucleophilic attack from alkyne group of 2-(phenylethynyl) aniline forms the first alkenyl carbocation and five-membered ring continuously activated by BF3·OEt2. With additional proton, the second alkenyl carbocation is given by attack from alkyne group of 2-(phenylethynyl)benzaldehyde together with novel six-membered ring. Then intramolecular Friedel−Crafts reaction and aromatization give rise to the second five-membered ring. The product dihydrobenzo[a]indeno[2,1-c] carbazole is finally obtained via 1,5-H shift, which is determined to be rate-limiting owing to the tension resulting from great structure bending required by long-distance shifting. The positive solvation effect is suggested by decreased absolute and activation energies in solution compared with in gas. These results are supported by Multiwfn analysis on FMO composition of specific TSs, and MBO value of vital bonding, breaking.
As privileged tricyclic nitrogen-containing hetero-cyclic motifs, carbazoles involve carbon skeleton of fluorene [1]. Polymers based upon 2,7-disubstituted carbazole function as electron-donating materials in organic photovoltaic devices [2]. They are also active compounds pharmaceutically and exist widely in alkaloid-based natural products [3-5], such as application in synthesis of carbazomycin A, calothrixin B, and staurosporinone. Its electron-rich and conjugated nature makes it prime as optoelectronic materials and organic fluorescent sensors with aggregation induced emission for detection of cyanide anion and Hg2+-protein binding [6,7]. Chen reported an indole-to-carbazole strategy under metal-free conditions for synthesis of carbazole derivatives demonstrating various biological activities including antimicrobial, anticancer and enzyme inhibition properties [8]. The stability and reactivity of carbazole are distinctive with a structure of two benzene rings fused to pyrrole ring [9]. Benzo[a]carbazoles are especially important among large varieties of carbazoles [10].
Benzo[a]carbazole is well known as core structure of bioactive products and key building blocks of organic materials. Paramasivam reported the influence of π-spacers in tuning the photovoltaic performance of benzocarbazole-based sensitizers for dye-sensitized solar cells [11]. Ivaniuk analyzed highly luminous sky-blue organic light-emitting diodes based on bis [(1,2)(5,6)] indoloanthracene emissive layer [12]. Furthermore, its derivatives are also widely used in pharmaceutical. For example, Ghanbari achieved direct synthesis of benzo [a] carbazoles by Palladium-catalyzed domino reaction and revealed photophysical properties to be agonists of human thrombopoietin receptor [13]. Kuo exploited dual character of benzoquinone as oxidizing agent and dienophile with powerful antitumor activities [14]. There is also Jeon’s cyanide-catalyzed imino-stetter reaction/Friedel−Crafts reaction sequence and Yuan’s Rhodium-catalyzed C−H activation/carbenoid insertion/Aldol-type cyclization [15,16]. Hence much interest has been focused on exploring efficient synthetic methods from materials readily available [17,18]. Such as the progress of Peng’s Gold (I)-catalyzed tunable domino cyclization of difunctional 1, 2-diphenylethynes and Rh(III)-catalyzed [5+1] annulation of 2-aryl-3-acyl-1H-indoles with a-diazo carbonyl compounds [19,20].
In spite of recent advancements for diverse heterocycles, the metal-free tandem protocols are relatively few as far as we know. Besides intramolecular aza-prins type reaction and stereoselective cascade alkynyl prins cyclization of Biswas group [21,22], there are unwavering commitments contributed by Chutia including temperature tunable synthesis of tetrahydro-4H-pyrrolo[3,2-c] quinolin-4-ones and tandem oxidation/iodolactonization of 2-O/N-tethered alkenyl benzaldehyde in this aspect [23,24]. Considering the merits of 2‑Alkynylaniline and 2-Alkynylbenzaldehyde, a breakthrough was Chutia’s Lewis acid (LA)-catalyzed tandem benzannulation and Friedel−Crafts reaction [25]. Although highly π-extended dihydrobenzo[a]indenocarbazole were yielded, many problems still puzzled and there was no report about detailed mechanistic study explaining the promotion of BF3·OEt2. How the vital imine intermediate is formed initially mediated by BF3·OEt2 and undergoes further activation? How alkenyl carbocation is obtained via two times of nucleophilic attack? What’s specific process of the last significant 1,5-H shift from intermediate of intramolecular Friedel−Crafts reaction? To solve these questions in experiment, an in-depth theoretical study was necessary for this strategy also focusing on the comparison of uncatalyzed paths and photophysical properties of products.
Optimized structures were obtained at M06-2X/6-31G(d) level of theory with GAUSSIAN09 [26]. In tests of popular DFT methods [27], M06-2X functional attained smaller standard deviation of difference between calculated value and experimental value in geometries than B3LYP including Becke's three-parameter hybrid functional combined with Lee−Yang−Parr correction for correlation [28,29]. The best compromise between accuracy and time consumption was provided with 6-31G(d) basis set on energy calculations. Also, M06-2X functional was found to give relatively accurate results for catalysed enantioselective (4 + 3), concerted [4 + 2], stepwise (2 + 2) cycloaddition and catalysed Diels−Alder reactions [30,31]. Together with the best performance on noncovalent interaction, M06-2X functional is believed to be suitable for this system [32-34]. The nature of each structure was verified by performing harmonic vibrational frequency calculations. Intrinsic reaction coordinate (IRC) calculations were examined to confirm the right connections among key transition-states and corresponding reactants and products. Harmonic frequency calculations were carried out at the M06-2X/6-31G(d) level to gain zero-point vibrational energy (ZPVE) and thermodynamic corrections at 333 K and 1 atm for each structure in acetonitrile.
The solvation-corrected free energies were obtained at the M06-2X/6-311++G(d,p) level by using integral equation formalism polarizable continuum model (IEFPCM) in Truhlar’s “density” solvation model [35-39] on the M06-2X/6-31G(d)-optimized geometries. As an efficient method obtaining bond and lone pair of a molecule from modern ab initio wave functions, NBO procedure was performed with Natural bond orbital (NBO3.1) to characterize electronic properties and bonding orbital interactions [40-42]. The wave function analysis was provided using Multiwfn_3.7_dev package [43] including research on frontier molecular orbital (FMO) and Mayer bond order (MBO).
The mechanism was explored for BF3·OEt2-facilitated benzannulation and Friedel−Crafts reaction of 2-(phenylethynyl) aniline 1 with 2-(phenylethynyl) benzaldehyde 2 leading to dihydrobenzo[a]indeno[2,1-c]carbazole 3 (Scheme 1). Illustrated by black arrow of Scheme 2, an imine intermediate a is formed through interaction between 2-alkynylaniline 1 and 2-alkynylbenzaldehyde 2 mediated by BF3·OEt2 via initial two steps. Subsequently, the imine group is continuously activated by BF3·OEt2 as corresponding Lewis acid, which undergoes nucleophilic attack by alkyne group of 1 facilitating the formation of alkenyl carbocation intermediate c and five-membered ring. Then c is further attacked by alkyne group of 2 yielding another alkenyl carbocation d and novel six-membered ring. Consecutively, an intramolecular Friedel−Crafts reaction ensues resulting in the formation of intermediate e, the aromatization of which gives rise to neutral intermediate f and the second five-membered ring. Finally, the desired product dihydrobenzo[a]indeno[2,1-c]carbazole 3 is obtained via significant [1,5-H shift] within intermediate f. The schematic structures of optimized TSs in Scheme 2 were listed by Figure 1. The activation energy was shown in Table 1 for all steps. Supplementary Table S1, Table S2 provided the relative energies of all stationary points. According to experiment, the Gibbs free energies in acetonitrile solution phase are discussed here.
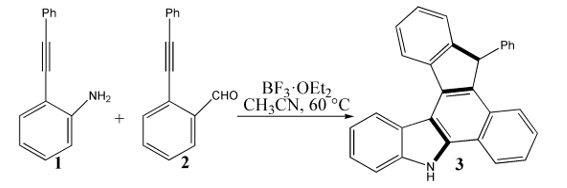
Scheme 1 BF3·OEt2-facilitated benzannulation and Friedel−Crafts reaction of 2-(phenylethynyl)aniline 1 with 2-(phenylethynyl)benzaldehyde 2 leading to dihydrobenzo[a]indeno[2,1-c]carbazole 3.
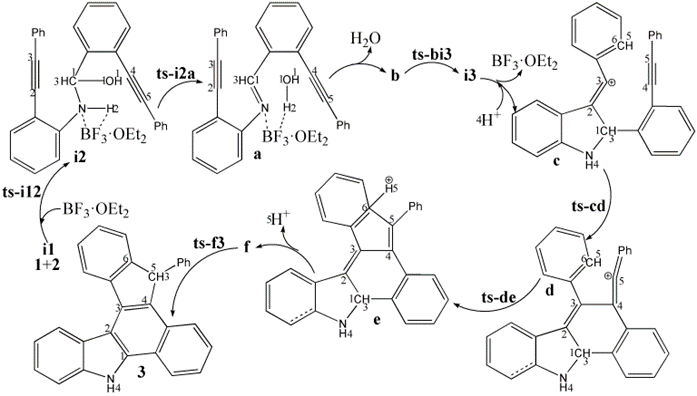
Scheme 2: Proposed reaction mechanism of BF3·OEt2-facilitated benzannulation and Friedel−Crafts reaction of 1 with 2 leading to 3. TS is named according to the two intermediates it connects.
| TS | ΔG≠gas | ΔG≠sol |
| ts-i12 | 18.5 | 16.7 |
| ts-i2a | 28.8 | 25.1 |
| ts-bi3 | 20.1 | 13.6 |
| ts-cd | 9.4 | 9.2 |
| ts-de | 6.6 | 8.5 |
| ts-f3 | 30.5 | 28.1 |
| n-tsi12 | 28.6 | 25.3 |
| n-tsi2a | 31.6 | 28.4 |
| n-tsbi3 | 26.9 | 21.1 |
Table 1: The activation energy (in kcal mol−1) of all reactions in gas and solvent
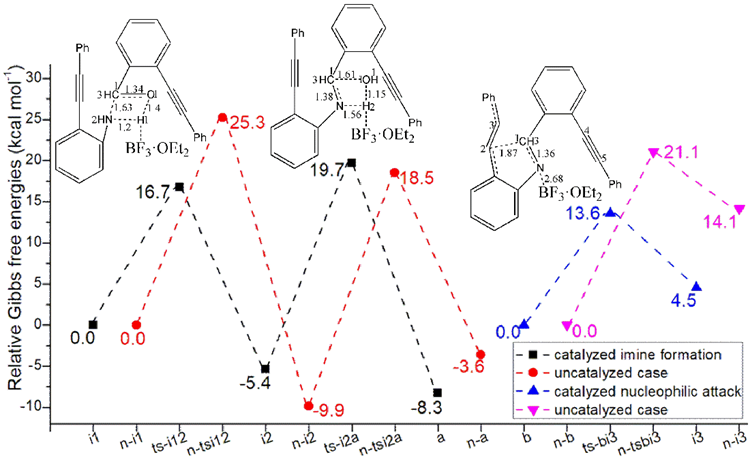
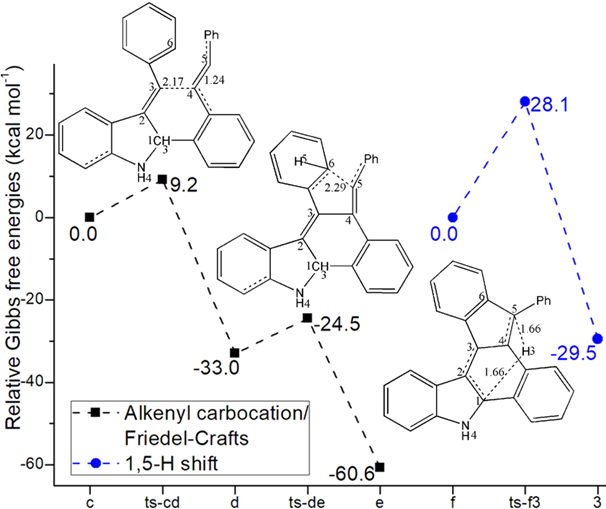
Figure 1: Relative Gibbs free energy profile in solvent phase starting from complex (a) i1, n-i1, b, n-b (b) c, f (Bond lengths of optimized TSs in Å).
| Species | ΔGgas | ΔGsol(CH3CN) |
| 1+2+LA | 0.00 | 0.00 |
| i1 | -18.98 | -12.10 |
| ts-i12 | -0.47 | 4.64 |
| i2 | -21.78 | -17.49 |
| ts-i2a | 7.00 | 7.58 |
| a | -19.85 | -20.36 |
| 1+2+LA-H2O | 0.00 | 0.00 |
| b | -6.17 | -2.55 |
| ts-bi3 | 13.96 | 11.04 |
| i3 | 0.34 | 1.98 |
| 1+2-H2O+H | 0.00 | 0.00 |
| c | 7.25 | 1.60 |
| ts-cd | 16.67 | 10.76 |
| d | -23.55 | -31.39 |
| ts-de | -16.96 | -22.90 |
| e | -44.87 | -59.02 |
| 1+2-H2O | 0.00 | 0.00 |
| f | -15.05 | -28.37 |
| ts-f3 | 15.41 | -0.29 |
| 3 | -44.09 | -57.85 |
| 1+2 | 0.00 | 0.00 |
| n-i1 | -13.81 | -7.83 |
| n-tsi12 | 14.80 | 17.42 |
| n-i2 | -14.20 | -17.70 |
| n-tsi2a | 17.36 | 10.71 |
| n-a | -8.06 | -11.42 |
| 1+2-H2O | 0.00 | 0.00 |
| n-b | -3.48 | -3.45 |
| n-tsbi3 | 23.41 | 17.63 |
| n-i3 | 12.56 | 10.69 |
Table S1. Calculated relative energies (all in kcal mol-1, relative to isolated species) for the ZPE-corrected Gibbs free energies (ΔGgas), Gibbs free energies for all species in solution phase (ΔGsol) at 333 K by M06-2X/6-311++G(d,p)//M06-2X/6-31G(d) method and difference between absolute energy.
| TS | ΔG≠gas | ΔG≠sol |
| ts-i12 (1318i) | 18.5 | 16.7 |
| ts-i2a (1131i) | 28.8 | 25.1 |
| ts-bi3 (308i) | 20.1 | 13.6 |
| ts-cd (386i) | 9.4 | 9.2 |
| ts-de (215i) | 6.6 | 8.5 |
| ts-f3 (1566i) | 30.5 | 28.1 |
| n-tsi12 (1426i) | 28.6 | 25.3 |
| n-tsi2a (1013i) | 31.6 | 28.4 |
| n-tsbi3 (411i) | 26.9 | 21.1 |
Table S2. The activation energy (local barrier) (in kcal mol−1) of all reactions in the gas, solution phase calculated with M06-2X/6-311++G(d,p)//M06-2X/6-31G(d) method.
| N···C1 | N···H1 | H1···O1 | C1···O1 | |
ts-i12 | 0.80 | 0.50 | 0.22 | 1.25 |
| N···C1 | N···H2 | H2···O1 | C1···O1 | |
| ts-i2a | 1.38 | 0.18 | 0.71 | 0.38 |
| C2···C1 | N···C1 | |||
| ts-bi3 | 0.24 | 1.12 | ||
| C4···C3 | C4···C5 | |||
| ts-cd | 0.32 | 1.79 | ||
| C6···C5 | ||||
| ts-de | 0.68 | |||
| C1···H3 | H3···C5 | |||
| ts-f3 | 0.41 | 0.40 |
Table S3. Mayer bond order (MBO) of typical TSs
3.1 BF3·OEt2-mediated imine formation/nucleophilic attack of alkyne
During initial two steps, BF3·OEt2 constantly functions as LA not only accepting lone pair electrons via B-N bond but forming H bond depending on F. The complex between 1, 2 and BF3·OEt2 is denoted as i1. The nucleophilic addition of 1 to 2 proceeds via ts-i12 in step 1 with the activation energy of 16.7 kcal mol−1 relative to the starting point i1
exothermic by -5.4 kcal mol−1 producing i2 (black dash line of Figure 1a). The transition vector containstwo parts in concerted modes that is nucleophilic attack of negative N to positive C1 along with cooperated transferring of H1 from N to O1 and carbonyl group C1-O1 stretching from double bond to single one (1.63, 1.2, 1.4, 1.34 Å) (Figure S1a). Obviously, i2 is stable with formal N-C1 single bond.
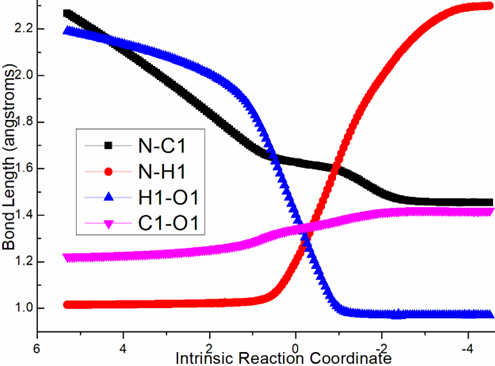
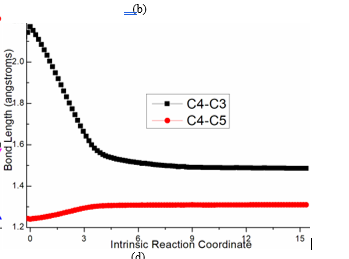
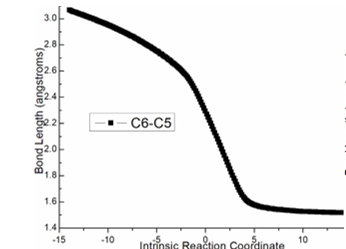
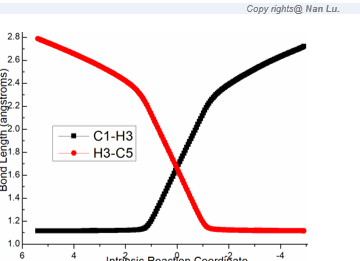
Figure S1. Evolution of bond lengths along the IRC for (a) ts-i12(b) ts-cd(c) ts-de (d) ts-f3 at M06-2X/6-311++G(d,p) level.
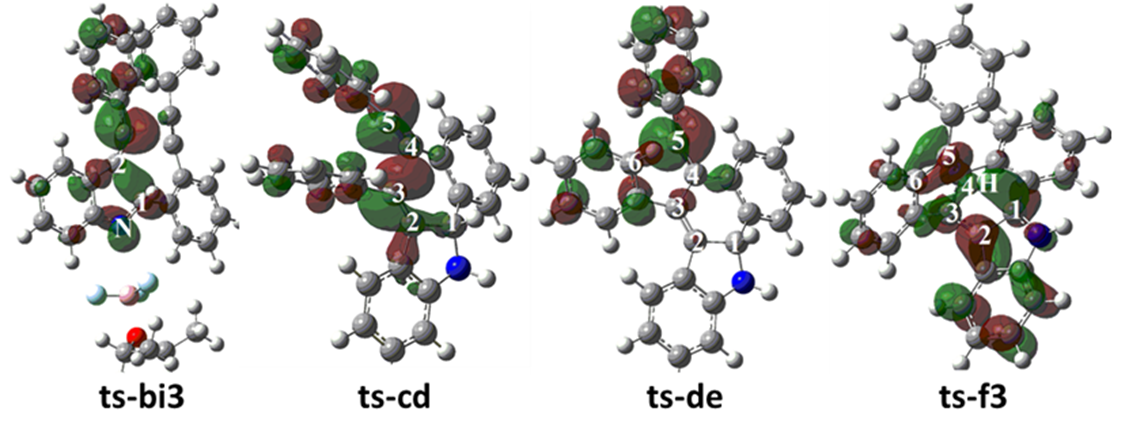
Figure S2. Highest Occupied Molecular Orbital (HOMO) of typical TSs. Different colors are used to identify the phase of the wave functions.
Then mediated by BF3·OEt2, the dehydration process occurs via ts-i2a as step 2 with activation energy of 25.1 kcal mol−1 exothermic by -8.3 kcal mol−1 giving intermediate a. The transition vector includes another H2 on N moving to O1 forming water molecule H1-O1-H2 as well as the consquent breaking of C1-O1 single bond, further shortening of N-C1 from single to double (1.56, 1.15, 1.61, 1.38 Å). Both N and C1 turn to be sp2 hybrid in resultant stable a with typical imine structure.
Subsequently, after the removal of water, the imine group N-C1 is continuously activated by BF3·OEt2 via B-N interaction in the similar intermediate b, which is taken as new starting point initiating nucleophilic attack from alkyne group of 1. This step 3 takes place via ts-bi3 with activation energy of 13.6 kcal mol−1 affording i3 endothermic by 4.5 kcal mol−1 (blue dash line of Figure 1a). According to the transition vector, as the negative alkyne C2 approaches positive C1, the imine C1-N double bond is elongated promoting π electron focusing on N and further cleavage of B-N bond (1.87, 1.36, 2.68 Å). Hence BF3·OEt2 is leaving from then on and the role of LA is ended in the following process. Once C2-C1 is formed, the first five-membered ring is obtained in i3.
3.2 Alkenyl carbocation formatiom/intramolecular Friedel−Crafts/1,5-H shift
An additional proton H4 bonded to negative N facilitates the formation of alkenyl carbocation intermediate c with positive charge on alkyne C3. This makes the next two steps from starting point c fairly easy. In step 4 c is further attacked by alkyne group of 2 via ts-cd with activation energy of 9.2 kcal mol−1 exothermic by -33.0 kcal mol−1 delivering intermediate
d (black dash line of Figure 1b). The transition vector corresponds to the approaching of negative C4 to positive C3 and C4-C5 tripple bond is weakened to double one (2.17, 1.24 Å) (Figure S1b). When C4-C3 single bond is completed, the stable d is yielded as the second alkenyl carbocation with novel six-membered ring. In addition, alkyne of 2 becomes accumulated diene with positive charge shifting to C5 ready for the vital Friedel−Crafts reaction closely afterwards.
The intramolecular Friedel−Crafts reaction happens via ts-de in step 5 with activation energy of 8.5 kcal mol−1 exothermic by -60.6 kcal mol−1 generating intermediate e rather stable. The transition vectorreveals remarkable attack from sp2 hybrid C6 of phenyl to positive C5 (2.29 Å) (Figure S1c). The resulting e is characterized by C6-C5 single bond, the second new five-membered ring and sp3 hybrid C6 with positively charged H5. Consecutively, the aromatization of e gives rise to neutral intermediate f wihtout H5 to accomplish the last step.
Finally, the significant [1,5-H shift] proceeds via ts-f3 in step 6 with activation energy of 28.1 kcal mol−1 relative to f yielding the desired product dihydrobenzo[a]indeno[2,1-c]carbazole 3 exothermic by -29.5 kcal mol−1 (blue dash line of Figure 1b). The detailed atomic motion is illustrated according to the transition vector about proton H3 transfer from C1 to C5 (1.66, 1.66 Å) (Figure S1d). Evidently, the energy of ts-f3 is elevated owing to the tension caused by great structure bending required by long-distance shifting. Therefore, 1,5-H shift of step 6 is determined to be rate-limiting for the whole process.
To highlight the idea of feasibility for changes in electron density and not molecular orbital interactions are responsible of the reactivity of organic molecules, quantum chemical tool Multiwfn was applied to analyze of electron density such as MBO results of bonding atoms and contribution of atomic orbital to HOMO of typical TSs (Table S3, Figure S2). These results all confirm the above analysis.
3.3 Comparison with uncatalyzed case
The reaction without BF3·OEt2 are also explored to investigate the promotion of LA. The complex between 1, 2 is denoted as n-i1. The nucleophilic addition of 1 to 2 proceeds via n-tsi12 in step 1 with the activation energy of 25.3 kcal mol−1 exothermic by -9.9 kcal mol−1 producing n-i2. The next dehydration occurs via n-tsi2a as step 2 with activation energy of 28.4 kcal mol−1 exothermic by -3.6 kcal mol−1 giving n-a also with typical imine structure of N-C1 double bond the same as that of a (red dash line of Figure 1a).From n-b, the nucleophilic attack from alkyne group of 1 takes place via n-tsbi3 in step 3 with activation energy of 21.1 kcal mol−1 affording n-i3 endothermic by 14.1 kcal mol−1 (magenta dash line of Figure 1a).
From kinetics, the barries of previous three steps are all increasedfor uncatalyzed process compared with BF3·OEt2-facilitated case. Furthermore, the relative energy of most stationary points are comparatively high and is not advantageous from the perspective of entire potential energy surface. The catalytic effect of BF3·OEt2 as LA was fully confirmed from two aspects of lowering activation energy and relative energies.
4 Conclusions
Our DFT calculations provide the first theoretical investigation on BF3·OEt2-facilitated benzannulation and Friedel−Crafts reaction of 2-(phenylethynyl)aniline with 2-(phenylethynyl)benzaldehyde. An imine structure is formed mediated by BF3·OEt2 constantly functioning as LA via initial two steps. The subsequent nucleophilic attack from alkyne group of 2-(phenylethynyl)aniline forms the first alkenyl carbocation and five-membered ring continuously under the influence of BF3·OEt2. With additional proton, the second alkenyl carbocation is given by attack from alkyne group of 2-(phenylethynyl)benzaldehyde together with novel six-membered ring. Then an intramolecular Friedel−Crafts reaction and aromatization give rise to the second five-membered ring. The desired product dihydrobenzo[a]indeno[2,1-c]carbazole is obtained via 1,5-H shift, which is determined to be rate-limiting with higher barrier than other five steps owing to the tension resulting from great structure bending required by long-distance shifting. The catalytic effect of BF3·OEt2 as LA is confirmed from both lowering activation energy and relative energies for previous three steps. The positive solvation effect is suggested by decreased absolute and activation energies in acetonitrile solution compared with in gas. These results are supported by Multiwfn analysis on FMO composition of specific TSs, and MBO value of vital bonding, breaking.
Supplementary data available: [Computation information and cartesian coordinates of stationary points; Calculated relative energies for the ZPE-corrected Gibbs free energies (ΔGgas), and Gibbs free energies (ΔGsol) for all species in solution phase at 333 K.
Conceptualization, Nan Lu; Methodology, Nan Lu; Software, Nan Lu; Validation, Nan Lu; Formal Analysis, Nan Lu; Investigation, Nan Lu; Resources, Nan Lu; Data Curation, Nan Lu; Writing-Original Draft Preparation, Nan Lu; Writing-Review & Editing, Nan Lu; Visualization, Nan Lu; Supervision, Chengxia Miao; Project Administration, Chengxia Miao; Funding Acquisition, Chengxia Miao. All authors have read and agreed to the published version of the manuscript.
This work was supported by National Natural Science Foundation of China (21972079) and Key Laboratory of Agricultural Film Application of Ministry of Agriculture and Rural Affairs, P.R. China.
The authors declare no conflict of interest.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
