
Case Report | DOI: https://doi.org/10.31579/2690-4861/216
1 Assistant Professor of Cardiology, Sri Jayadeva Institute of Cardiovascular Sciences & Research, Bangalore
2 Professor of Cardiology, Sri Jayadeva Institute of Cardiovascular Sciences & Research, Bangalore.
*Corresponding Author: Arnab Ghosh Chaudhury, Ex-assistant professor of cardiology, sri jayadeva institute of cardiovascular sciences & research., bangalore. (Currently working as consultant cardiologist at Vivekananda hospital, Durgapur)
Citation: Arnab Ghosh Chaudhury, Prabhavathi Bhat, C N Manjunath. (2022) The Squeezed Heart-A Case Report J. International Journal of Clinical Case Reports and Reviews. 11(2); DOI: 10.31579/2690-4861/216
Copyright: © 2022 Arnab Ghosh Chaudhury, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 05 April 2022 | Accepted: 15 April 2022 | Published: 04 May 2022
Keywords: pompe’s disease; biventricular hypertrophy; glycogen storage disease
Pompe’s disease is an autosomal recessive disorder caused by inherited deficiency of α-1,4-glucosidase (acid maltase), a lysosomal enzyme. Patients usually die in the first year of life from cardio-respiratory failure due to massive left ventricular hypertrophy. We report a case of 3-month-old boy presented with fatal infantile onset Pompe’s disease.
Glycogen storage disease type II or Pompe’s disease is a rare hereditary error of carbohydrate metabolism in which excessive quantities of glycogen accumulate in the heart, muscle and other tissues [1,2].Pompe’s disease is characterized by infantile onset muscular weakness, hypotonia, and enlargement of the heart and liver, followed by progressive cardiorespiratory failure [2]. We report a case of 3-month-old boy presented with progressive respiratory distress who had infantile onset Pompe’s disease.
A 3-month-old male infant born out of consanguineous marriage presented with respiratory distress for 2 weeks which worsened over last 3 days. He had significant history of diaphoresis, feeding difficulties and poor weight gain since birth. On examination, patient had tachypnoea, tachycardia, chest retractions, massive hepatomegaly, generalised hypotonia. Cardiovascular examination revealed loud S1 due to tachycardia, normal S2, no murmur. As the baby was extremely sick, emergency echocardiography was done. 2D echocardiography in parasternal long axis and short axis view revealed massive biventricular hypertrophy with slit like left ventricular cavity as if it is squeezed by thick ventricular wall (FIGURE 1A & 1B).

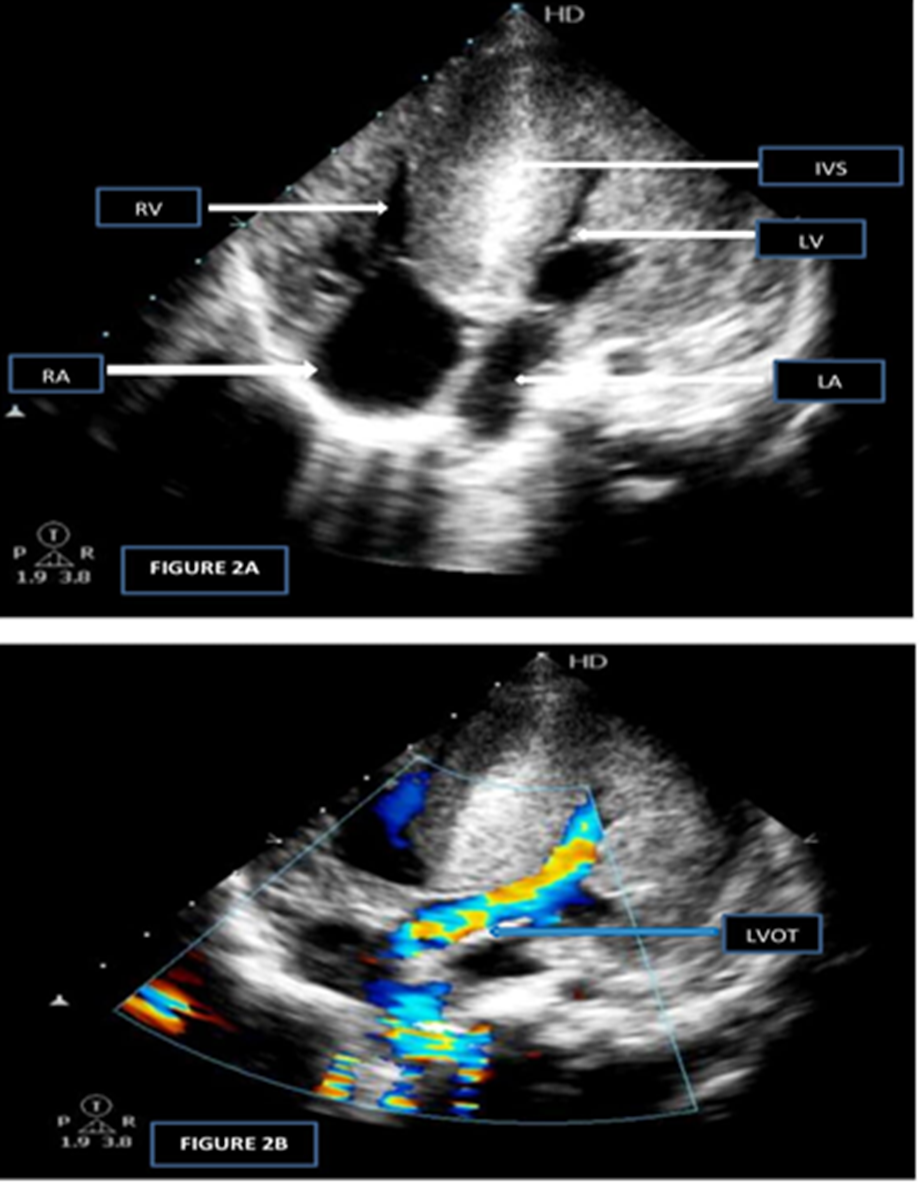
Apical four chamber view revealed biventricular hypertrophy with normal sized atria (FIGURE 2A). Apical five chamber view showed normal left ventricular outflow tract with normal aortic valve (FIGURE2B).
Subcostal view revealed normal sized atria with normal inter atrial septum (FIGURE 3A). Suprasternal view ruled out any evidence of coarctation of aorta (FIGURE 3B).

In view of suspected diaphragmatic palsy patient was shifted to paediatric neurology ICU. Biventricular hypertrophy without left ventricular outflow tract (LVOT) obstruction was the provisional morphological diagnosis. Differential diagnosis of storage disorders (Type II, III or IV glycogen storage disease), HCM variants, PRKAG2 mutation, Danon disease, infant of diabetic mother was thought of. In view of high clinical suspicion of Pompe’s disease, α-1, 4-glucosidase leucocyte enzyme assay was sent. The enzyme was significantly low: 1.76nmol/h/mg protein (normal range, 3.3–14.5 nmol/h/mg protein) confirming the diagnosis of Pompe’s disease (Type II glycogen storage disease). Final diagnosis was infantile onset Pompe’s disease presented with generalised muscle weakness, hepatomegaly, and cardiomyopathy with biventricular hypertrophy. Unfortunately, the infant died of respiratory failure before enzyme replacement therapy could be started.
Pompe’s disease or type II glycogen storage disease occurs due to deficiency of lysosomal acid α-1, 4-glucosidase enzyme [3]. It results in lysosomal glycogen accumulation principally in cardiac, skeletal, and smooth muscle cells. Pompe’s disease has autosomal recessive inheritance, so history of consanguineous marriage is very frequent as in our case and affected babies are usually males. Pompe’s disease can be of two types: Infantile onset or juvenile adult onset. Infantile onset Pompe’s disease presents with generalised hypotonia, hepatomegaly, hypertrophic cardiomyopathy and cardio-respiratory failure before first birthday [4]. Juvenile and adult-onset variants have less clinical severity, usual presentation is progressive proximal muscle weakness without cardiomyopathy. Differential diagnosis includes Danon disease (LAMP2 mutation), PRKAG2 mutation, Fabry’s disease. Danon disease (LAMP2 mutation) is a lysosomal glycogen storage disease with normal acid maltose whereas acid maltose is essentially low in Pompe’s disease [5]. Danon disease has X-linked dominant inheritance, childhood onset, presents with cardiomyopathy, skeletal myopathy, mental retardation [5]. Death occurs usually in 2nd to 3rd decade. PRKAG2 mutation has autosomal recessive inheritance, usual presentation is hypoglycaemia, cardiomyopathy and early death in infancy.Fabry’s disease occurs due to alpha-galactosidase A deficiency [6]. It is a x-linked-recessive disease usually presents with renal, cardiac, nervous system and gastrointestinal involvement [6]. But usual clinical manifestation of Fabry’s disease occurs at 30-45 years of age, it is not clinically apparent in infancy. In our case mother was nondiabetic. So maternal diabetes related ventricular hypertrophy of baby was ruled out. Infantile onset hypertrophic cardiomyopathy was also thought of which is exceedingly rare.
Successful treatment is available for Pompe’s disease in the form of enzyme replacement therapy [Recombinant human GAA: alglucosidase alfa, (Myozyme)]. Timely administration of enzyme replacement can prevent fatality and regress organomegalies to a great extent. To conclude, in every case of floppy infant with hepatomegaly and biventricular hypertrophy without LVOT obstruction, Pompe’s disease should be suspected, α-1, 4-glucosidase in leucocytes should be assessed as confirmatory test, prompt enzyme replacement therapy must be started if Pompe’s disease is diagnosed.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
