
Review Article | DOI: https://doi.org/10.31579/2690-4861/268
Oncology department (Radiotherapy Department) of Zhe Jiang Hospital, Hangzhou, Zhejiang, China
*Corresponding Author: Zhi-Bing Wu, No. 1229, Gudun Road, Xihu District, Hangzhou City, Zhejiang Province, China
Citation: Lei L., Li-T. Chen, Lai J., Zhi-B. Wu (2022). The Role of PFKFB3 In the Tumor Microenvironment and Its Therapeutic Potential. International Journal of Clinical Case Reports and Reviews. 12(2); DOI:10.31579/2690-4861/268
Copyright: © 2022 Zhi-Bing Wu, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 19 October 2022 | Accepted: 24 October 2022 | Published: 16 November 2022
Keywords: glucose metabolism; warburg effect; 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase; glucose metabolism ; metabolic reprogramming
Energy metabolic reprogramming has recently been recognized as an important hallmark of tumor metabolic biology, and tumor cells can be manipulated to obtain rapid growth, immune evasion and apoptosis, which play an important role in tumor development. Enhanced glycolysis is the main source of energy required for tumorigenesis, development and metastasis in the tumor microenvironment, and Warburg suggested that tumor cells preferentially choose glycolysis as the main pathway to provide energy even under aerobic conditions, but the mechanisms and causes of this metabolic alteration remain unclear. PFKFB3 is an important activator of glycolysis and plays an important role in carcinogenesis, including Tumor cytogenesis, metastasis, drug resistance and alteration of the tumor microenvironment. In this review, we summarize the role of PFKFB3 in different tumor types and its effects on the tumor microenvironment, as well as the signaling pathways through which PFKFB3 acts in tumors, suggesting PFKFB3 as a potential anti-tumor therapeutic target.
The "Warburg effect", first proposed by the German scientist Otto Warburg in the early 1920s [1,2], It is that tumor cells preferentially choose glycolysis as the main metabolic pathway to provide energy even under adequate oxygen conditions. The level of glycolytic flux is regulated by different mechanisms, including three key enzymes, namely hexokinase, phosphofructokinase-1 (PFK-1) and pyruvate kinase [3]. One of the key steps is the conversion of fructose-6-phosphate (F6P) to fructose-1,6-phosphate(F1,6P2). 6-phosphofructo-1-kinase, for which F-1,6-BP, ADP, and AMP are metabolic activators. Its activity is regulated by adenosine triphosphate (ATP), adenosine diphosphate (ADP), and fructose-2,6-phosphate (F-2,6-BP) [4]. The level of Fru-2,6-P2 in tumor cell glycolysis is mainly regulated by PFKFB3, a hallmark of malignancy that is susceptible to regulation by isoforms characterized by high kinase/phosphatase ratios. The PFKFB3 gene has the highest kinase/phosphatase activity in the PFKFB gene family and is used to maintain high glycolytic efficiency [5,6]. (Figure 1)
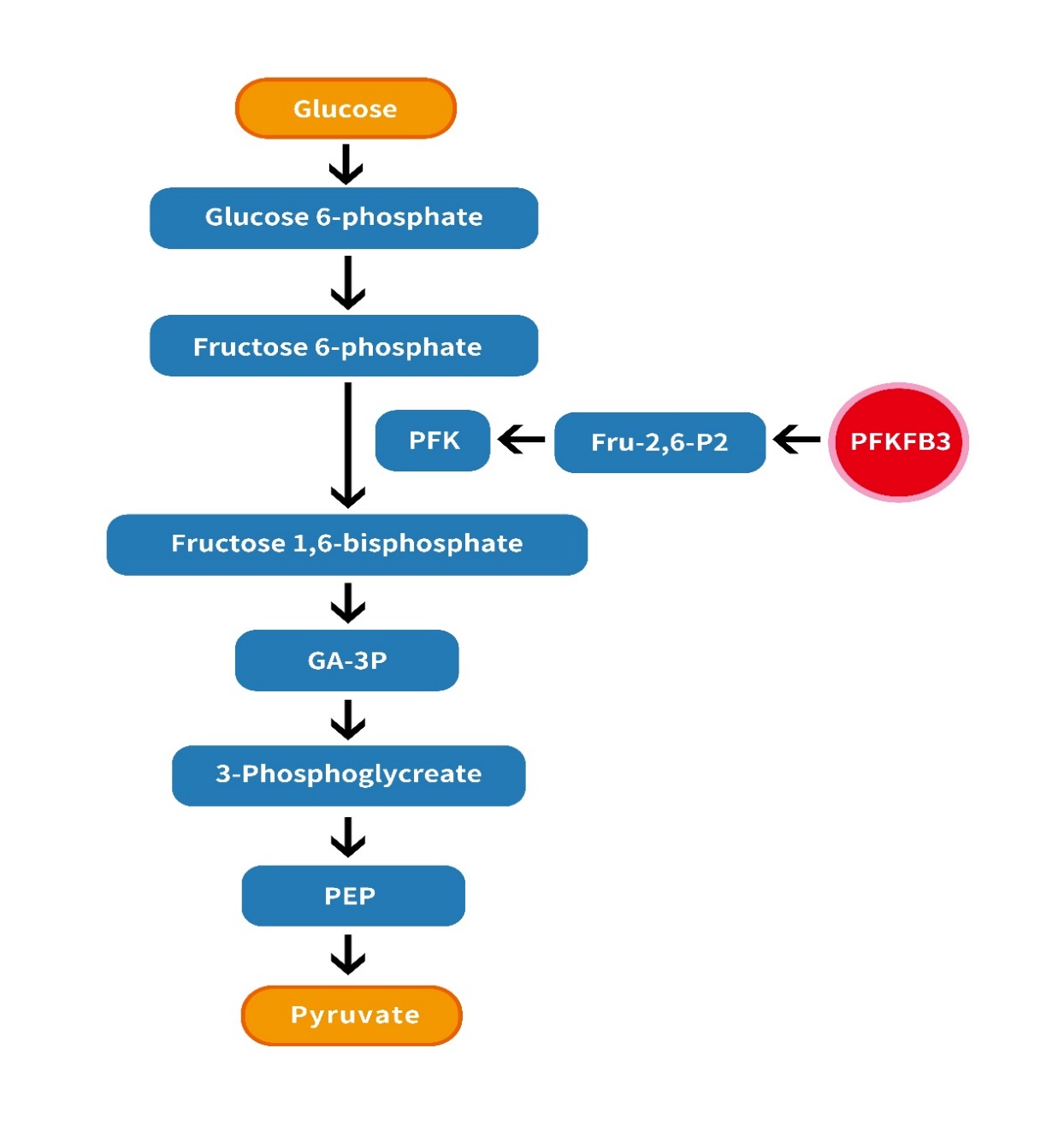
Figure1: The role of PFKFB3 in glycolysis is illustrated by the regulation of PFK-1: PFKFB3 promotes glycolysis by activating F-2,6-BP.
The PFKFB gene consists of four different isozymes (PFKFB1-4) [7], which are highly homologous, among which the PFKFB3 gene is located on the short arm of chromosome 10p14-15 [8] and was first isolated in the bovine brain library [9], in which the pfkfb3 gene consists of 19 exons including 7 variable regions and 12 constant regions The kinase/bisphosphatase activity is 700-fold higher than that of the isozyme and facilitates the synthesis of Fru-2,6-P2 [10,11]. This PFKFB3 protein has two distinct functional domains: the C-terminal domain of the diphosphatase activity of the enzyme, which degrades F2,6P2 to F6P and inorganic phosphate (Pi), and this structural domain can be selectively spliced to produce six isoforms from UBI2K1 to UBI2K6. the N-terminal domain is responsible for the synthesis of F2,6P2 from F6P and ATP. the PFK-2/FBPase2 bifunctional enzyme is the gene product enzyme of PFKFB3 and is regulated by covalent modifications. the C-terminal structural domain can be phosphorylated at Ser461 by different protein kinases [12]. (Figure2, Figure3)
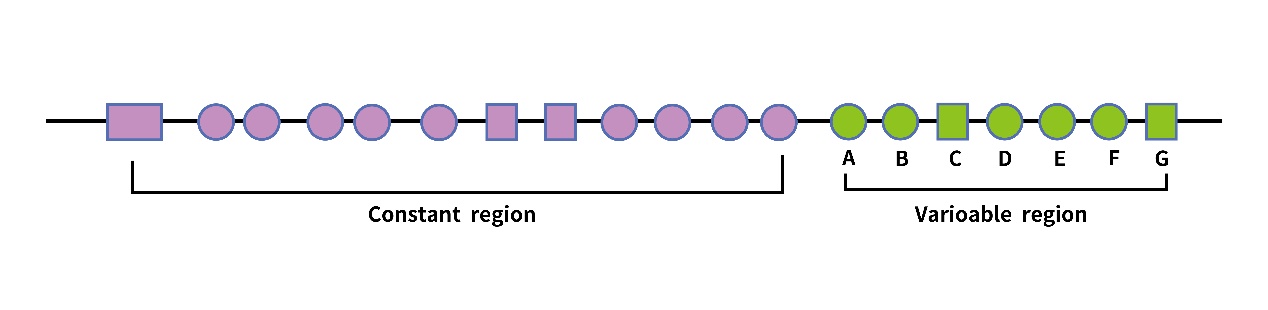
Figure2: PFKFB3 gene structure:The PFKFB3 gene contains 19 exons, which are divided into a constant region and a variable region. The variable region contains seven exons from A-G.
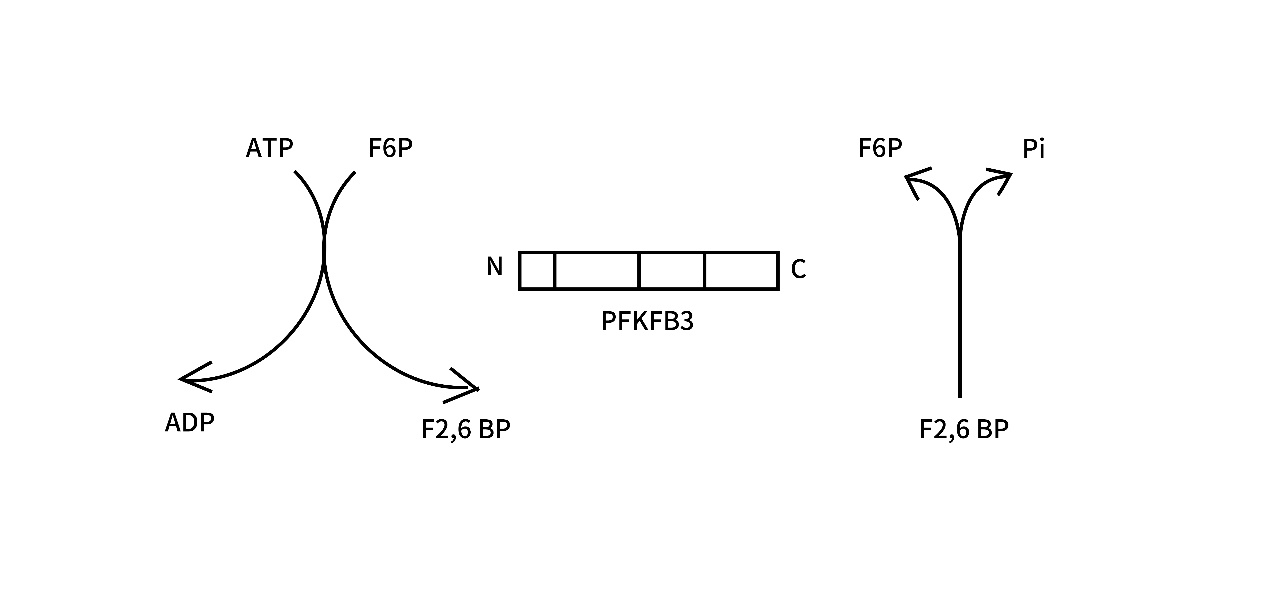
Figure3: PFKFB3 protein structure: The PFKFB3 protein has two homodimeric subunits, each containing an N-terminal kinase domain and a C-terminal phosphatase domain. the N-terminal catalyzes the production of F2,6P2 and ADP from F6P and ATP, which highly promotes the glycolytic pathway. the C-terminal dephosphorylates F2,6P2 to produce F6P and Pi.
1.Targeting PFKFB3 in immune cells
Warburg effect is associated with the regulation of immune cell metabolism, and both immune cells and tumor cells are considered as highly motile cells that preferentially select glycolysis for energy supply, and by exploiting the possibility of regulating the metabolism of cancer cells and antitumor immune cells by metabolic differences between different cells, glycolysis can be used as a metabolic checkpoint for cancer immunotherapy. Chen [13] et al. found that monocytes in the tumor microenvironment significantly increased the glycolytic flux of cells in the paracancerous region of HCC (hepatocellular carcinoma). The activation of glycolysis induced PD-L1 expression on cells in the peritumoral region of HCC, thereby attenuating cytotoxic T lymphocyte infiltration. The key glycolytic enzyme PFKFB3 can be upregulated by soluble factors such as hyaluronic acid. PFKFB3 not only acts as a cellular glycolytic switch, but also mediates increased PD-L1 expression through activation of the nuclear factor kappa B signaling pathway. PFKFB3/CD68 infiltration in hepatocellular carcinoma paraneoplastic tissues was negatively correlated with overall survival of HCC patients and was an independent prognostic factor for survival of HCC patients. The role of PFKFB3 in T lymphocyte-mediated immune-related antitumor immunotherapy is still poorly investigated. Zhang [14-16] et al. found that PFKFB3 inhibitor PFK-158 increased infiltrating CD8+ and CD4+ T cells and decreased infiltrating Th17 cells in melanoma mice. Glycolytic reprogramming is associated with immune activation responses and immune tolerance in the tumor environment and is a potential target for future immune-based anticancer therapies.
2. PFKFB3 and ECs
Tumors are rich in blood vessels with irregular morphology, which provide nutrition for tumor development and metastasis and are associated with poor prognosis, and pathological angiogenesis is one of the characteristic hallmarks of tumors [17,18]. Under conditions such as hypoxia, vascular endothelial growth factor (VEGF) in vascular endothelial cells can stimulate neovascularization by regulating PFKFB3 expression [19,20], and regulate directed migration of vascular endothelial cells to promote neovascularization and endothelial migration. [21].
In contrast to conventional anti-angiogenic therapies aimed at reducing angiogenesis, it is the normalization of the characteristically disorganized tumor vascular system to improve blood perfusion, which may reduce hypoxia and increase drug accessibility. Normalized blood vessels may also resist the shedding of cancer cells in the primary tumor, which may reduce tumor metastasis. A recent study found that inhibition of PFKFB3 in melanoma tumor vascular endothelial cells (TEC) induced normalization of tumor blood vessels, thereby reducing tumor angiogenic activity and contributing to increased sensitivity to antitumor drugs. Improving vascular permeability by reducing VE-calmodulin endocytosis thereby reducing angiogenic activity [22,23]. By reducing NF-κB signaling, TECs also decrease the expression of cancer cell adhesion molecules and inhibit metastasis of cancer cells [24]. Therefore, targeting EC metabolism by PFKFB3 may provide novel therapeutic perspectives for anti-angiogenic therapy and inhibition of tumor growth.
3. PFKFB3 and autophagy
In 1963 Christian de Duve elucidated the mechanism of the process from yeast to cultured cell lines into mice,
The concept of autophagy was first proposed [25], which consists of five distinct phases: initiation, vesicle nucleation, vesicle elongation, vesicle fusion and material degradation, regulated by ancient and highly conserved genes that act during stresses such as oxidative stress, hypoxia and nutrient deprivation [26,27]. Autophagy is an organelle-dependent process that transports substances to lysosomes for degradation, ensures the cell cycle and provides energy to the cell. Autophagy presents a double-edged role in tumors depending on the tumor microenvironment: it can be both a tumor suppressor mechanism and a promoter of tumor cell survival. For example, in renal cell carcinomas, PFKFB3 accumulation in the cytoplasm promotes ATP production and thus inhibits autophagy in renal cell carcinoma (RCC) cells, whereas nuclear localization of PFKFB3 is associated with enhanced autophagy in tumor cells [28]. Self-renewal of breast cancer stem cells is an important process in the resurrection of breast cancer after metastatic dormancy, PFKFB3 plays an important conditional role in breast cancer metastatic recurrence, Alyssa La Belle Flynn observed an inverse relationship between PFKFB3 expression and autophagy, disruption of autophagy timing drives PFKFB3 to be able to drive dormant BCSC and metastatic lesion recurrence The phenotypic shift from a metastatic dormant state to tumor recurrence from high PFKFB3low autophagy to low PFKFB3 high autophagy was observed, and the detection of PFKFB3 and autophagy expression status could predict tumor recurrence and the response status of tumor patients to treatment[29].
4. PFKFB3 and Tumor drug resistance
The mortality rate of hepatocellular carcinoma is extremely high worldwide, largely due to the lack of effective and durable therapeutic measures. [30] Sorafenib is the first generation of targeted therapy for hepatocellular carcinoma and is considered to be of benefit to patients, however, because of the early onset of sorafenib resistance, most patients do not receive a long-term benefit. Li et al. showed that aspirin combined with sorafenib could induce death of sorafenib-resistant cancer cells without associated hepatotoxicity and inflammatory responses by inhibiting PFKFB3 to reduce glycolytic flux to make mitochondrial permeability, and by making metabolites such as lactate decrease to activate associated apoptotic signals. It is shown that PFKFB3 overexpression dominates the resistance process of sorafenib in hepatocellular carcinoma and can be controlled by inhibiting PFKFB3 expression through aspirin as a target. In hepatocellular carcinoma sorafenib enhances glucose uptake and lactate export in hepatocellular carcinoma cells [31]. It has been shown that glycolysis inhibitors such as inhibition of PFKFB3 can greatly inhibit the growth of sorafenib-resistant cells to increase glycolytic flux and enhance glycolysis [32,33]. Silencing of PFKFB3 enzyme has a synergistic effect with sorafenib [34,35,36]. Inhibition of glycolysis by specific molecules or targeting key enzymes of glycolysis is an effective strategy to attenuate sorafenib resistance.
Cisplatin is the first widely used chemotherapeutic agent in solid tumors such as head and neck tumors, lung cancer, bladder and reproductive tumors, where the mechanism of action is mainly to induce DNA damage in tumor cells leading to cell cycle arrest and induction of apoptosis. Demonstrated that PFKFB3 has a key role in protecting tumor cells from chemotherapeutic drug-induced apoptosis. Cisplatin-induced DNA damage induces PFKFB3 acetylation at lysine 472 (K472), and acetylation at Lys472 of PFKFB3 protein contributes to the cytoplasmic accumulation of PFKFB3 by weakening the interaction between input protein α5 and PFKFB3. This increases the phosphorylation of PFKFB3 at the Ser461 site. This is known to be associated with enhanced glycolytic activity. Cisplatin-induced DNA damage stimulates PFKFB3 Lys472 acetylation in an ATM-dependent manner, promoting enhanced glycolysis and protecting cells from cisplatin-induced apoptosis. This is critical for the cellular response to DNA damage caused by chemotherapeutic agents. A novel mechanism for acetylation-mediated PFKFB3 accumulation in the cytoplasm regulating cisplatin antitumor was revealed, and a potential strategy for chemotherapy by targeting PFKFB3 was proposed[37].
5. regulation mechanism
The mechanisms regulating PFKFB3 are different for different environments and different regulatory factors(Table1). For example, progesterone, stress stimulation or insulin trigger a dual mechanism to ensure that glycolysis increases phosphorylation and activation of PFKFB3 enzymes in acute environmental situations [38,39]. Activation of different kinases in hypoxic, ischemic or stressful microenvironments may be critical for cell survival and therefore represent a protective mechanism for cells in stressful states or hypoxic environments.HIF-1, AMPK and other kinases are components of a concerted cellular response to maintain energy homeostasis in hypoxic, stressful or ischemic microenvironments [39, 40,41].PFKFB3 is subject to Demethylation [42] or ROS-mediated S glutathionylation [43] is regulated in cancer cells, resulting in tumor cells primarily utilizing the pentose phosphate pathway for metabolism rather than the glycolytic pathway, which allows for ROS detoxification. p53 represses PFKFB3 gene expression to increase glucose passage through the pentose phosphate pathway to increase nucleotide production, thereby promoting nucleotide biosynthesis in response to DNA damage [44].

Table 1
The oncogenic Ras signaling pathway is a central regulator of glucose metabolism for cancer and can regulate PFKFB3 activity. Ras in glioblastoma inhibits glycolysis by reducing pfkfb3 gene expression through inhibition of hypoxia-inducible factor 1 α (HIF-1α) [39]. [45] Constitutive HER2 expression increases PFKFB3 expression and glucose metabolism in breast cancer cells. Hypoxia, progesterone, and estradiol can induce PFKFB3 expression through the interaction of HIF-1, progesterone receptor (PR), and estrogen receptor (ER) with their shared response elements located in the pfkfb3 promoter. It was shown that the circadian rhythm-driven transcription factor 'CLOCK' can bind to the pfkfb3 promoter at the 'E-box' locus to increase pfkfb3 transcription in cancer cells, PFKFB3 inhibition significantly delays the growth of implanted human tongue cancer cells in mice only at certain time points within the circadian cycle [46]. This finding suggests the importance of time-based PFKFB3 inhibition in cancer therapy. The major signaling pathways involved in PFKFB3 regulation were shown to be in (Figure4)
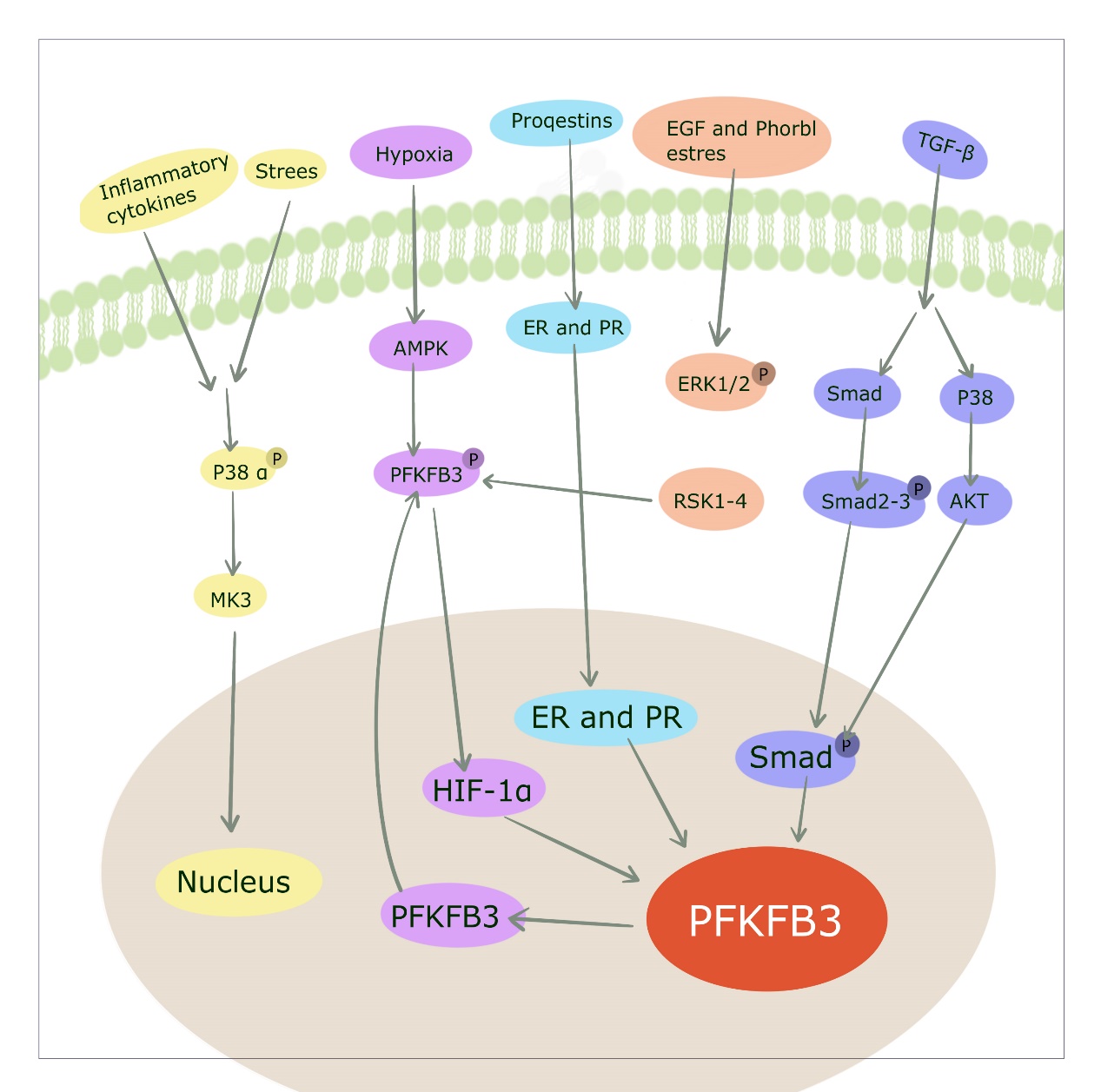
Figure4: Numerous signaling pathways that regulate PFKFB3. [1] Progesterone, estradiol and hypoxia induce the transcription factors PR, ER and HIF to bind to their response elements at the PFKFB3 promoter, respectively. [2] Stimuli such as stress increase PFKFB3 production through the P38/MK3 pathway. eGF acts through the ERK1/2 (extracellular signal-regulated kinase)/RSK1-4 (ribosomal S6 kinase) pathway, and [3] the TGF-β/Smad pathway regulates PFKFB3 expression
6. PFKFB3 role in tumor proliferation and invasion
Atsumi et al. showed that PFKFB3 mRNA G1/S cell cycle phase was induced to promote tumor cell proliferation [47]. In line with these findings, Calvo et al. found a significant decrease in tumor growth rate by silencing PFKFB3siRNA in HeLa adenocarcinoma cervical cancer cells [48]. Furthermore, Yalcin et al. showed that PFKFB3 knockdown induced cell cycle arrest at G1/S in HeLa cells [49]. The pro-proliferative role of PFKFB3 in the tumor cell cycle was confirmed.
PFKFB3 expression was also negatively correlated with many proteins involved in epithelial-mesenchymal transition (EMT). Gu M et al. showed that knockdown of PFKFB3 in nasopharyngeal carcinoma cells revealed up-regulated expression of E-cadherin and down-regulation of Vimentin and N-cadherin levels [50]. In addition, it has been shown that PFKFB3 siRNA transfection reduced Snail expression while upregulating E-cadherin levels in pancreatic cancer cells [51]. Our previous study showed that PFKFB3 upregulated expression in gastric cancer cells correlated with gastric cancer EMT. aberrant PFKFB3 expression and tumor invasion and metastasis.
With the accumulation of knowledge about the role of PFKFB3 in tumor metabolic reprogramming, there has been a strong interest in PFKFB3 inhibitors in antitumor therapy(Table2). To date, [52,53] the best-known PFKFB3 inhibitor is 3PO, a 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one, also known as 3PO, synthesized by Clem et al. 3PO inhibits a variety of tumor cells by inhibiting recombinant PFKFB3, decreasing 3PO inhibits a variety of tumor cells by inhibiting recombinant PFKFB3, reducing glucose uptake and decreasing cytoplasmic levels of Fru-2,6-P and ATP, thereby attenuating tumor cell activity. PFKFB3 expression was increased in HER2-positive breast cancer, and PFKFB3 expression was associated with poorer progression-free and metastasis-free survival in breast cancer patients [45].PFK15 showed greater inhibitory activity against PFKFB3, rapidly induced apoptosis in transformed cells, displayed adequate pharmacokinetic properties, inhibited glucose uptake and 20 Promoted Lewis in syngeneic mice lung cancer growth in syngeneic mice and exerted antitumor effects in thymus-free mice against three human cancer xenograft models [54].
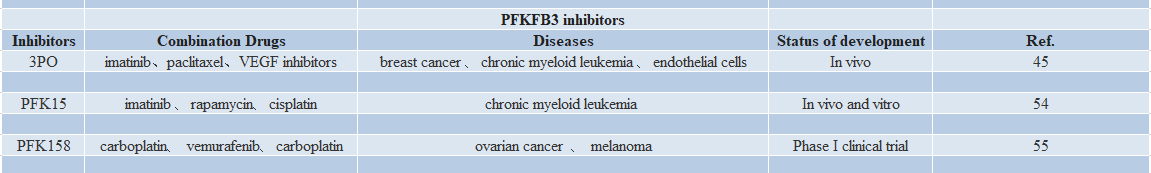
Table 2
PFK15 is the only PFKFB3 inhibitor used in phase I clinical trials, and a phase I clinical trial of PFK158 in patients with advanced solid malignancies was completed in 2016, confirming the efficacy of PFK158 in a variety of advanced solid malignancies. PFK158 has been shown to be safe and protective in a variety of advanced solid tumors, demonstrating its important clinical utility as monotherapy or in combination with other targeted agents (clinicaltrials.gov #NCT02044861) [55,56,57].
In this review, we summarize the current knowledge on the role of PFKFB3 in tumor cell metabolism and the mechanisms of PFKFB3 action. The intriguing links between PFKFB3 and tumors, and between ECs and immune cells, suggest that PFKFB3 is a potential target for tumor therapy. However, to date, no PFKFB3 inhibitors have been approved for the treatment of cancer patients. 3PO is a potent inhibitor of PFKFB3, but poor water solubility makes this compound clinically unusable, and the recent emergence of nanotechnology-based drug delivery vehicles has the ability to formulate a variety of hydrophobic anticancer agents, including 3PO, thus showing potential to enhance anticancer effects when used in vivo. PFKFB3 represents a promising target for tumor therapy. However, to date, no PFKFB3 inhibitors have been approved for the treatment of cancer patients. 3PO is a potent inhibitor of PFKFB3, but poor water solubility has prevented the clinical use of this compound. Other potent and selective PFKFB3 inhibitors, such as PFK15 and PFK-158, are in clinical trials in patients with advanced tumors. Nanotechnology-based drug delivery vehicleswere formulated into various hydrophobic anticancer agents, including 3PO, thus showing potential to enhance anticancer effects when used in vivo.The relationship between PFKFB3 as a metabolic regulatory switch and tumor immunity suggests that targeting tumor metabolism and targeting immunity to treat tumors provides new insights.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
