
Review Article | DOI: https://doi.org/10.31579/2641-0419/136
1 Department of Cardiology, Zhongda Hospital, School of Medicine, Southeast University 87 Dingjiaqiao, Nanjing, P.R.China 210009
*Corresponding Author: Yongjun Li, Genshan Ma, Department of Cardiology, Zhongda Hospital, School of Medicine, Southeast University 87 Dingjiaqiao, Nanjing, P.R.China 210009
Citation: Xi Chen., Mingming Yang., Pengfei Zuo., Rui Zhang., Zaixiao Tao., Genshan Ma., Yongjun Li., (2021) The role of non-coding RNA in Cardiac Repair and Regeneration after Myocardial Infarction. J. Clinical Cardiology and Cardiovascular Interventions, 4(6); Doi:10.31579/2641-0419/136
Copyright: © 2021 Yongjun Li, Genshan Ma, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 02 February 2021 | Accepted: 25 February 2021 | Published: 02 March 2021
Keywords: long non-coding RNAs; myocardial infarction; cardiac repair
Myocardial infarction (MI), one of the cardiovascular diseases (CVDs) with high incidence and mortality rate, seriously endangers human health. The poor ways of fully repairing and regenerating the infarcted myocardium may have an impact on people's life quality, therefore scientists have devoted continuously to exploring the way of myocardial repair after MI so as to strive for a better prognosis of these patients. In recent years, non-coding RNAs (ncRNAs) have been identified and become one of the exciting fields of research in the development of CVDs. In a wide range of areas, more and more research has found that ncRNAs play important roles in myocardial repair. This review mainly introduces some strategies for myocardial repair and the role or mechanism of microRNA (miRNA), long non-coding RNA (lncRNA), circular RNA (circRNA) and circRNA/lncRNA-miRNA-mRNA regulatory axis in the repair of myocardial tissue after MI, in order to build a better understanding and find new therapeutic targets for MI.
BM-MSCs : bone marrow derived mesenchymal stem cells
CVDs : cardiovascular diseases
circRNA : circular RNA
Col Ⅰ, Ⅲ : collagen Ⅰ, Ⅲ
CTGF : connective tissue growth factor
CXCR4 : C-X-C chemokine receptor type 4
DNMT : DNA (cytosine-5-)-methyltransferase
ERK1/2 : extracellular signal-regulated kinase 1/2
EGFL7 : epidermal growth factor-like domain 7
HIF-1α : hypoxia inducible factor 1α
lncRNA : long non-coding RNA
MAPK : mitogen-activated protein kinase
MI : myocardial infarction
miRNA, miR : microRNA
miPSC : mouse-induced Pluripotent stem cell
mTOR : mammalian target of rapamycin
ncRNAs : non-coding RNAs
PI3K : phosphoinositide 3 kinase
PDGF : Platelet derived growth factor
PCFL : Primary cutaneous follicular lymphoma
RISC : miRNA-induced silencing complex
TGF-β : transforming growth factor β
3' UTR : 3' untranslated region
VEGF-A : vascular endothelial growth factor-A
Myocardial infarction (MI), a cardiovascular disease with high morbidity and mortality in the world, is caused by blockage of coronary arteries, leading to apoptosis or necrosis of cardiomyocytes induced by continuous ischemia and hypoxia1. Evidence has shown that many cardiomyocytes are lost when MI occurs. Fibrous repair, also called scar repair is thought to be the main way of repair after MI because cardiomyocytes regeneration capacity is limited [2]. However, a large amount of scar tissue seriously affects myocardial systolic and diastolic function, which greatly increases the incidence of adverse events followed by MI [3]. Therefore, finding more ideal ways and strategies for myocardial repair after MI has become the research hotspot and difficulty of many scientists.
At present, it is unanimously believed that the most ideal way to repair damaged myocardium is to promote the regeneration of myocardium as a substitute for apoptotic cardiomyocytes4. In addition, studies have also focused on promoting angiogenesis in the infarct area5, inhibiting myocardial cell apoptosis [6], inflammatory response [7], excessive fiber and collagen deposition [8]. Although scientists have been constantly exploring, so far it is still at the level of simple animal or cell experiments, and more molecular mechanisms and clinical treatments need to be explored.
It is reported that about 2% of the human genome is composed of protein-coding regions. Most transcripts are non-coding RNAs (ncRNAs), mainly including microRNA (miRNA, miR), long non-coding RNA (lncRNA), and circular RNA (circRNA) [9]. The ncRNAs have been confirmed to play important roles in occurrence and development of various diseases through different molecular mechanisms10. MiRNA, together with some proteins, forms the miRNA-induced silencing complex (RISC), which acts on the 3' untranslated region (3' UTR) of the target mRNA and then degrade it or inhibit it's translation. Overall miRNA may participate in a variety of biology functions and control various cellular processes by negatively regulating gene expression11. Different from miRNA, lncRNA and circRNA mainly act as a "sponge" or endogenous competitor molecule of miRNA to affect the miRNA’s function expression which finally regulate the occurrence and development of various human diseases [12]. In recent years, it has been reported that miRNA, lncRNA and circ-RNA play significant roles in repairing damaged myocardium [13], so this paper mainly reviews the role and mechanism of lncRNA, miRNA, circRNA separately and the regulatory networks of lncRNA/circRNA-miRNA-mRNA in tissue repair after MI for the purpose of providing some new ideas and targets for the treatment of MI (figure1).
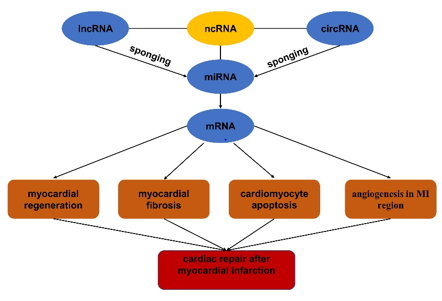
NcRNAs in myocardial regeneration
The loss of a large number of cardiomyocytes when someone have a MI seriously affects the prognosis. Although scientists have been searching for better therapies to repair injured myocardial tissue, to date regenerate cardiomyocytes is recognized as the most ideal treatment method for repair of damaged myocardium [4], for example transplanting stem cell to induce cardiomyogenic differentiation, activating and inducing resident cardiac progenitor cells or cardiomyocytes to re-enter the cell proliferation cycle [14-16]. And increasing evidence indicate that the activating of cardiomyocytes proliferation and the survival rate of stem cells transplanted into the infarct area are controlled by the microRNA network [4,13].
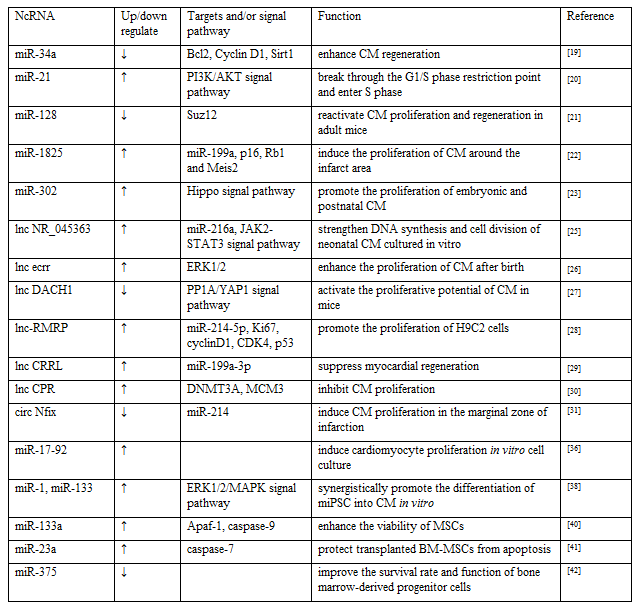
Discussion on whether the cardiomyocytes could regenerate has always been a contentious issue. The more accepted view is that heart of the adult mammalian, as a terminal differentiation of organs, is unable to regenerate after myocardial injury [2]. However, in recent years, some studies have pointed out that the heart of newborn mammals maintain a short-term regenerative capacity after birth, and then this ability gradually get lost [17,18]. Therefore, reactivation of cardiac cell proliferation is a key strategy for inducing cardiac regeneration in adults. Yang et al. stated that the level of miR-34a in the heart of mice after birth was low, and rose to adult levels soon, Antagonist of miR-34a enhanced endogenous repair and regeneration following MI partly by regulating cell cycle factors and apoptotic factors, including Bcl2, Cyclin D1 and Sirt1 [19]. MiR-21 has been reported to break through the G1/S phase restriction point and enter S phase to promote cardiac stem cell proliferation through the activation of phosphoinositide 3 kinase (PI3K) /AKT signaling pathway [20]. In early postnatal mice, myocardium-specific overexpression of miR-128 inhibited the proliferation of cardiomyocyte. In contrast, knocking out miR-128 can reactivate cardiomyocyte proliferation and regeneration in adult mice, which is partly achieved by targeting Suz12 in the heart to regulate cell cycle-related genes [21]. Pandey et al. discovered the expression of miR-199a could be regulated by transfected miR-1825 in animal model, which in turn downregulates the expression of p16, Rb1 and Meis2 to induce the proliferation of cardiomyocytes around the infarct area [22]. The miR302-367 cluster has been observed to have huge potential in promoting the proliferation of embryonic and postnatal cardiomyocytes, and the mechanism maybe lies in regulating targeting multiple kinases via Hippo signal transduction pathway, then Wang’s group innovatively injected hyaluronic acid gel wrapped miR-302 mimics to myocardial infarction site of mouse, then led to cardiomyocyte proliferation in the marginal area [23]. Similarly, Pierluigi group found that administration of lipid formulations of hsa-miR-199a-3p and hsa-miR-590-3p immediately after MI in mice resulted in marked reduction of infarct size and persistent recovery of cardiac function [24]. In recent years, some scientists have made innovative use of bioengineering to deliver target molecules into specific cells in the heart, so as to exert their best effect as well as pushed miRNA therapeutics a step forward toward clinical application. But these techniques are still immature and remain some safety issues, more experiments are needed to improve them.
It has been widely accepted that lncRNA and circRNA can regulate the process of cell proliferation and differentiation. lncRNA NR_045363, a highly conserved lncRNA, is mainly expressed in cardiomyocytes. Wang et al. noticed that over-expression of NR_045363 could strengthen DNA synthesis and cell division of neonatal cardiomyocytes cultured in vitro, The mechanism may be that NR_045363 interacts with miR-216a, thereby regulating JAK2-STAT3 pathway to promote cardiomyocyte proliferation [25]. Chen et al. reported that lnc RNA ecrr enhanced the proliferation of cardiomyocyte after birth and the recovery of function following MI through the extracellular signal-regulated kinase 1/2 (ERK1/2) dependent mechanism [26]. Cai et al. claimed that LncDACH1 is gradually upward in the heart after birth, while knocked out LncDACH1 and silenced adenovirus-mediated endogenous LncDACH1 in vivo conditional, the proliferative potential of cardiac myocytes was activated in both young and adult mice, further mechanism research revealed that LncDACH1 induced heart regeneration after birth and ischemic heart by regulating PP1A/YAP1 signal [27]. Yan et al. found that the lncRNA-RMRP/miR-214-5p axis regulated the expression levels of Ki67, cyclinD1 and CDK4 by directly targeting p53, ultimately promoted the proliferation of H9C2 cells induced by hypoxia [28]. Contrary to the proliferative effect of lncRNA-RMRP, human-derived lncRNA CRRL is a new negative regulator of myocardial cell regeneration after MI. CRRL suppressed myocardial regeneration by acting as an endogenous miR-199a-3p sponge [29]. LncRNA CPR, known as cardiomyocyte proliferation regulator, has been verified in animal experiments to interacted directly with DNMT3A and MCM3 promoter CpG island methylation, restrained MCM3 expression and involved in Inhibition of cardiomyocytes proliferation [30]. Emerging evidence suggested that circRNAs may play fundamental roles in promoting or inhibiting cardiac regeneration. For instance, circRNA Nfix (circNfix) is regulated by a super-enhancer and overexpressed in adult hearts of humans, rats and mice. In vitro study, the expression of circNfix did cut down in circNfix knockout cell lines from HL-1 mouse cardiomyocytes, meanwhile it was interesting that proliferation of cardiomyocytes significantly increased. Subsequently, in animal model of MI, the circNfix was down-regulated by injecting AAV9-shcircnfix or AAV9-shNC in the area around the infarction, as expected, 14 days after MI, the down-regulation of circNfix induced cardiomyocyte proliferation in the marginal zone of infarction by 2%-4%, and the mechanism by which circNfix controlled cardiomyocyte proliferation may be to promote Y-box binding protein 1 ubiquitin degradation by acting as a miR-214 sponge [31], the result showed the great potential of circRNA to promote cardiomyocyte regeneration and effective cardiac repair, But at present, there are not many related researches, and a lot of exploration is needed.
Stem cell therapy has always been a research hotspot in the field of MI. More and more scholars are devoted to repairing ischemia-induced myocardial injury with stem cells [32]. It has been confirmed that stem cell transplantation can differentiate into cardiomyocytes and promote the repair of injured myocardium. The mechanism may be related to ncRNAs. Recently exploitation of miRNA in stem cells for post-MI has to be attempted. Some scholars have begun to artificially interfere with the content of certain RNAs in stem cells, which may more effectively promote the differentiation of stem cells into cardiomyocytes [33,34]. For example, in vitro experiments, Yang et al. mentioned that the conditioned medium produced by mesenchymal stem cells (MSCs) had the ability to promote the proliferation of cardiomyocytes, which is enhanced after the suppression of miR-21. At the same time, an in vivo study confirmed that after inhibition of miR-21 expression, the ability of MSCs transplanted to the infarcted site for treating MI is improved [35]. A similar study have found that overexpression of miR-129 may promote the differentiation of bone marrow derived mesenchymal stem cells(BM-MSCs) into myocardial-like cells by activating the Wnt signaling pathway, while miR-148a induced MSCs to differentiate into myocardium through targeted regulation of DNA (cytosine-5-)-methyltransferase 1(DNMT1), Chen et al. conducted genetic studies using miR-17-92 gene knockout and transgenic mice, as well as in vitro cell culture, which results showed that the members of the 17-92 cluster are necessary to induce cardiomyocyte proliferation [36]. Transplantation of miR-1 transfected MSCs raised the survival rate of transplanted cells and the rate of cardiogenic differentiation, which is conducive to the repair of infarct injury [37]. In addition, it has been reported that miR-1 and miR-133 are highly expressed in myocardial tissue and participate in that process of myocardial formation and development in embryonic stage, whereas the separate upregulation of miR-1 or miR-133 have no obvious promoting effect in the process of differentiation into cardiomyocytes induced by mouse-induced Pluripotent stem cell (miPSC). Only when overexpression of miR-1 and miR-133 at the same time, can they synergistically promote the differentiation of miPSC into cardiomyocytes in vitro, this effect is mainly achieved by increasing the phosphorylation level of in ERK1/2/ mitogen-activated protein kinase (MAPK) pathway [38], which suggests that there may be some synergistic effect between miRNAs. An in vitro experiment found LncRNA Bravehearl transfection can effectively promote the differentiation of BM-MSCs into cardiomyocyte-like cells. However, it is reported that the efficacy of stem cell transplantation is still not ideal. One of the important factors is the low survival rate and differentiation efficiency of stem cells in the local infarct microenvironment in vivo, and their ability to differentiate into cardiomyocytes is extremely limited, so exploring to promote the survival and differentiation of stem cells in the infarct microenvironment efficiency has become a difficult problem to be solved.
In addition to regulating myocardial differentiation, miR-133 has also been reported to inhibit the apoptosis of MSCs under hypoxic conditions [39]. Dakhlallah et al. found that transfection of miR-133a mimics can enhance the viability of MSCs in the heart of MI by reducing the mRNA expression of Apaf-1 and caspase-940. MiR-23a has been stated to protect BM-MSCs from apoptosis under hypoxic conditions by regulating caspase-741. An in vitro study have shown that the miR-21 can reduce apoptosis, promote proliferation and differentiation of MSCs [35], Garikipati et al. emphasized that the negative regulation of IL-10 on miR-375 can improve the survival rate and function of bone marrow-derived progenitor cells in ischemic myocardium, and improve the repair after MI [42]. Another animal study described that transplantation of MSCs with overexpressing miR-1 into the infarcted area can promote cardiogenic differentiation by improving the survival rate of transplanted cells [37], although above studies have shown that ncRNAs do have a certain effect on improving the microenvironment of myocardial infarction area and the survival rate of stem cells, but it is far from enough. With the development of stem cell therapy research, scholars have gradually found that some transplanted stem cells improve heart function mainly through their paracrine function, there are a large number of miRNAs carried by paracrine exosomes, can be taken up by surrounding cells and participate in the repair of damaged myocardium[43]. A new type of cell-free therapy based on exosomes secreted by stem cells can be considered as a safe and effective alternative to stem cell therapies for cardiovascular disease [44] (Table 1).
NcRNAs in regulation of myocardial fibrosis
it is well known that cardiomyocytes cannot be completely regenerated following MI. The main repair method is scar repair, which is manifested as myocardial fibrosis and specifically includes the proliferation of myocardial fibroblasts as well as excessive deposition of extracellular matrix [45]. In the early post-MI period, fibrosis in the infarcted area is a favorable pathologic repair process, conducive to healing of scar tissue and prevention of ventricular rupture. However, with the extension of infarction time, fibrosis in the peripheral area of the infarction will seriously affect myocardial contractility and diastolic function, which is not beneficial to repair of cardiac function [46]. More and more scholars have proposed reasonable regulation of myocardial fibrosis after infarction and inhibition of late myocardial fibrosis is a valuable strategy for myocardial tissue repair, plenty of evidence indicate that miRNAs are a new type of myocardial fibrosis regulator that can regulate the proliferation activities of cardiac fibroblasts [47]
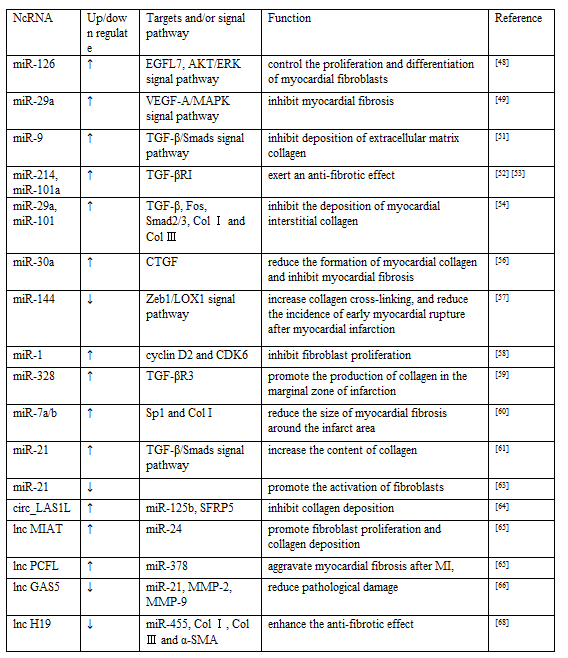
Some miRNAs have been shown to inhibit myocardial fibrosis, for example, miR-126 may control the proliferation and differentiation of myocardial fibroblasts by raising the expression of epidermal growth factor-like domain 7(EGFL7) and activating the AKT/ERK signaling pathway [48], while miR-29a targets vascular endothelial growth factor-A (VEGF-A)/MAPK signaling pathway inhibited myocardial fibrosis [49]. Platelet derived growth factor (PDGF) is a low molecular weight cytokinin that stimulates the growth of connective tissue and other tissue cells [50]. PGDFR-β was confirmed to be a target molecule of miR-9, overexpression of miR-9 disturbed myocardial fibrosis by targeting PDGFR-β. Previous studies have shown that the transforming growth factor β (TGF-β)/Smads signal transduction pathway plays a significant role in the process of regulating myocardial fibrosis. After the onset of myocardial infarction, the content of TGF-β1 in myocardial tissue increased, and the signal transduction pathway of TGF-β/Smads was activated rapidly, which promoted the expression of downstream genes, mainly including type Ⅰ and type Ⅲ collagen(Col Ⅰ, Col Ⅲ), resulting in continuous deposition of extracellular matrix collagen [51]. MiR-214 [52] and miR-101a [53] have been reported to directly down-regulate the expression of TGF-βRI in cardiac fibroblasts, thereby inhibiting the TGF-β signaling pathway and exerting an anti-fibrotic effect. Recently, an interesting study concluded that controlling intermittent aerobic exercise can help repair myocardial tissue and improve cardiac function after MI. In that second week after successful preparation of the AMI rat model, the mice began sustained aerobic exercise, and as a result, it was observe that the progression of myocardial fibrosis and scar formation was delayed, further studies have revealed that aerobic exercise induced high expression of miR-29a and miR-101 and regulates the level of fibrotic factors including TGF-β, Fos, Smad2/3, Col Ⅰ and Col Ⅲ, thereby inhibiting the deposition of myocardial interstitial collagen [54]. Chen et al. transfected the vector carrying the miR-30a gene into the heart of AMI model rats, and after 4 weeks it was found that highly expressed miR-30a directly binds to the 3'-UTR of connective tissue growth factor (CTGF) after four weeks. CTGF is a mitogen secreted by vascular endothelial cells [55]. In vitro experiments have shown that CTGF could stimulate the proliferation of myocardial fibroblasts and mediate tissues repair under pathological conditions. miR-30a inhibited the expression of CTGF post MI, then reduced the formation of myocardial collagen and inhibited myocardial fibrosis [56]. He et al. knocked out the miR-144 gene and established an AMI rat model, It was found that the deletion of miR-144 may increase collagen cross-linking, and reduce the incidence of early myocardial rupture after myocardial infarction via the Zeb1/LOX1 signal axis [57], but it is unclear whether it has a long-term effect. MiR-1 has been shown to negatively regulate fibroblast proliferation. MiR-1 has been reported to partially inhibit fibroblast proliferation mediated through targeted inhibition of cyclin D2 and CDK6 [58].
In addition, some miRNAs have been confirmed to promote myocardial fibrosis. Among them, miR-328 could promote the production of collagen in the marginal zone of infarction by targeting TGF-βR3 [59]. It was reported that overexpression of miR-7a/b reduced the size of myocardial fibrosis around the infarct area by decreasing the expression of Sp1 and Col I [60]. miR-21 has been confirmed to be mainly present in myocardial fibroblasts and is closely related to myocardial fibrosis after infarction. There are some scholars support the view that miR-21 could regulate the proliferation and differentiation process of fibroblasts, mainly through TGF-β/Smads signal transduction pathway. Liang et al. found that TGF-β1 and miR-21 were up-regulated, whereas TGF-βR3 was down-regulated in the border zone of mouse hearts in response to MI. Raising the expression of miR-21 could increase the content of collagen, at least in part by down-regulate TGF-βR3, on the contrary, the expression of TGF-βR3 can inhibit the expression of miR-21 in fibroblasts, reduce the production of collagen, can also make the TGF-β1 channel inactivation by lowering the expression of TGF-β1 and p-Smad3. Such a reciprocal loop between miR-21 and TGF-βR3 in myocardial fibrosis is worth exploring further [61]. However the specific regulatory role of miR-21 in myocardial fibrosis, the current research results are inconsistent, Chen et al. thought that inhibiting the expression of miR-21 instead promotes the activation of fibroblasts [62]. The specific regulatory role of miRNA-21 in myocardial fibrosis remains somewhat controversial, which may be related to the expression of miR-21 in different periods of MI and multiple regulatory mechanisms. However, it is worth noting that whether the positive or negative action of miRNA, including miR-21, reasonable regulation of miRNA expression levels and promotion of mutual cooperation or antagonism among miRNAs can regulate fibroblast proliferation and move toward a favorable pathological repair process to improve the prognosis of patients with MI.
With the continuous development of studies, the regulatory levels of circRNA and lncRNA are believed to be closely related to myocardial fibrosis. CircRNA and lncRNA have been proved to be generally acted as miRNA sponges, and play a regulatory role in circRNA-miRNA or lncRNA-miRNA modes. A recent study demonstrated that circ_LAS1L inhibited its activity by adsorbing miR-125b, thereby promoting SFRP5 expression and inhibiting collagen deposition [62]. MIAT, a kind of lncRNA, known as myocardial infarction-related transcript, was recently indicated that could promote fibrosis and regulate myocardial fibrosis after infarction which specifically promotes fibroblast proliferation and collagen deposition by down-regulating the expression of miR-24 [64]. Primary cutaneous follicular lymphoma (PCFL) has been proved to be a new type of profibrotic lncRNA, overexpression of PCFL can aggravate myocardial fibrosis after MI, and heterozygous knockout of PCFL can reverse this change, further mechanism exploration found that PCFL has the function of miR-378 sponge, which alleviated the inhibition of miR-378 on GRB2 [65]. Zhang et al. established the rat model of AMI by subcutaneous injection of isoproterenol, and observed the effect of lncRNA growth arrest specificity 5 (GAS5) on AMI rats, which showed that the cardiac function of the siRNA GAS5 group was significantly improved when compared with the model group, the myocardial tissue pathological damage was reduced, and the myocardial cell apoptosis rate was reduced. And they supposed underlying mechanism may be related to the up-regulation of miR-21 and the regulation of serum levels of MMP-2 and MMP-9 after MI [66]. lncRNA H19 has been thought to be an extracellular matrix regulator[67]. H19 knockout has been shown to enhanced the anti-fibrotic effect of miR-455, decreased the expression of CTGF, and further reduce the synthesis of fibrosis-related proteins(Col Ⅰ, Col Ⅲ and α-SMA) [68]. The above research indicated that circRNA and lncRNA play important roles in myocardial fibrosis, and generally regulate the expression level of miRNA in the form of miRNA sponge to regulate the proliferation of fibroblasts and other activities (Table 2).
NcRNAs in inhibition of cardiomyocyte apoptosis
Cardiomyocyte apoptosis is the most typical and intuitive pathological response after MI. When MI occurs, there is a large amount of cardiomyocyte necrosis in the area of infarct and a large number of cardiomyocytes in the marginal zone of the infarct undergo apoptosis6. The proliferation of cardiomyocytes was limited and could not regenerate completely to replace apoptotic cardiomyocytes, which seriously damaged the cardiac function of patients with AMI [69]. Therefore, inhibition of cardiomyocyte apoptosis induced by hypoxia is one of the therapeutic strategies for MI.
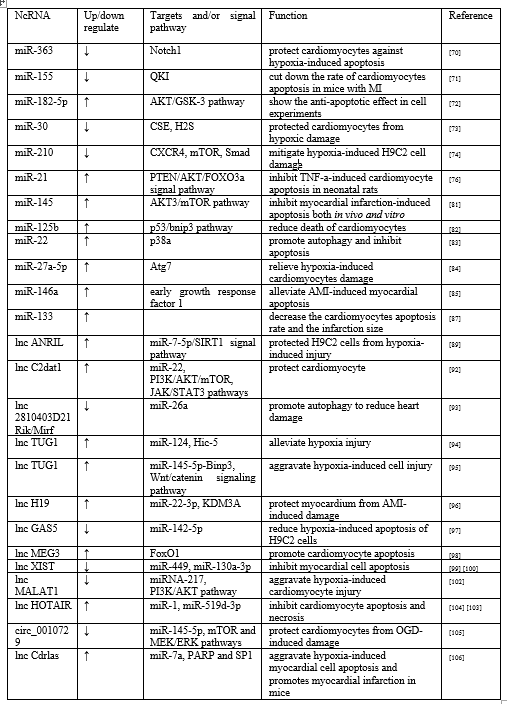
Increasing number studies have shown that various miRNAs can participate in the regulation mechanism of cardiomyocytes apoptosis. It has been reported that Inhibition of miR-363 could protect cardiomyocytes against hypoxia-induced apoptosis by promoting expression of Notch1 and activating it’s signaling pathway [70]. Inhibiting the expression of miR-155 also cut down the rate of cardiomyocytes apoptosis in mice with MI by targeting RNA-binding protein quaking (QKI), significantly narrowed infarction size [71]. Yao et al found that overexpression of miR-182-5p could inhibit the content of PTEN and enhance the AKT/GSK-3 signaling pathway to show the anti-apoptotic effect in cell experiments [72]. In vivo and vitro studies have shown that the over-expression of miR-30 family members reduced the expression of cysteine c-lyase (CSE) and the production of H2S, in turn aggravated the myocardial cell injury induced by hypoxia. In contrast, the down-regulation of the full miR-30 family protected cardiomyocytes from hypoxic damage [73]. Feng et al. found that C-X-C chemokine receptor type 4 (CXCR4) can be negative regulated by miR-210. Down-regulation of miR-210 mediated the expression of CXCR4, and then activated Smad and mammalian target of rapamycin (mTOR) signaling pathways to mitigate hypoxia-induced H9C2 cell damage, which provided a new regulatory axis for the expression of miR-210 after MI [74].
In studying the role of miRNA in the early stage of AMI, Ceviker et al. compared the miRNA expression profile in 6 hours after MI with the non-infarct area in rats. The results showed that the expression of miR-21 was significantly down-regulated in the infarct area and up-regulated in the marginal area of the infarct [75]. Subsequently Wang et al. found that increased expression of miR-21 could inactivated the PTEN/AKT/FOXO3a signaling pathway and inhibit TNF-a-induced cardiomyocyte apoptosis in neonatal rats [76]. Liu et al. found a positive feedback relationship between Hypoxia inducible factor 1α (HIF-1α) and miR-21 by regulating the PTEN/AKT signaling pathway. HIF-1α-miR-21 feedback plays an important role in inhibiting apoptosis during hypoxia. It is also reported that miR-1 has the potential to protect cardiomyocytes [77]. Xue et al. innovatively encapsulated miR-1 inhibitors into a dendritic nanocarrier coupled with myocardial-targeted at1 peptide, targeted injection into the myocardial infarction area, and found that the number of apoptotic cells in the dead marginal area was significantly reduced which shows the prospect of early treatment of MI [78]. Xu et al. further found that miR-1 and miR-21 have a synergistic protective effect on hypoxia-induced cardiomyocyte apoptosis, which was the advance on single miRNA study and needed more research [79].
Autophagy is an evolutionarily conserved process of intracellular degradation. It maintains intracellular homeostasis by removing damaged proteins and organelles to produce ATP. Autophagy can protect cell viability and reduce the infarct size following MI, subsequently, alleviate left ventricular remodeling under mild ischemia [80]. Yan et al. revealed that miR-145 induced autophagy via the AKT3/mTOR signaling pathway to inhibit myocardial infarction-induced apoptosis both in vivo and vitro [81]. Similarly miR-125b-rich exosomes downregulate autophagy level through the p53/bnip3 signaling pathway to reduce death of cardiomyocytes [82]. Li et al. determined that miR-22 regulates cardiomyocyte autophagy and apoptosis by targeting p38a both in animal and cell experiments [83]. MiR-27a-5p also has been shown to relieve hypoxia-induced cardiomyocytes damage because of Atg7-mediated autophagy and apoptosis [84]. There is some new evidence indicated that stem cells and exosomes secreted by them have great potentials in regulating apoptosis of cardiomyocytes. Pan et al. claimed that exosomes derived by adipose-derived stem cells that modified by miR-146a could alleviate AMI-induced myocardial apoptosis by down-regulating early growth response factor 1 [85]. Another study showed that transplanted BM-MSCs paracrine VEGF that can regulate the expression of miR-23a, miR-92a and other miRNAs, and which has exerted anti-apoptotic effect on myocardial cells after MI [86]. It has been proved that BM-MSCs with a high expression of miR-133 are implanted into the infarction area of rats. It can promote the survival of BM-MSCs, also decrease the cardiomyocytes apoptosis rate and the infarction size [87]. In recent years, it is reported that there are many ncRNAs in exosome secreted by stem cells. Among several cardio-protective miRNAs of MSC exosomes. Luther et al. found that miR-21a-5p was the most abundant. MiR-21a-5p-mediated cardio-protective effects is achieved by down-regulating PDCD4, FasL, PTEN and Peli1 and other pro-apoptotic gene [88]. Certainly, even at the highest content of miR-21a-5p, the role of other miRNAs in regulating myocardial cell apoptosis cannot be ignored. In the future, more unknown miRNAs in exosomes and their miRNA regulation networks should be further explored, even including circRNA and lncRNA.
In recent years, it has been shown that LncRNAs plays significant roles in regulating ischemia and hypoxia-induced cardiomyocyte death, lncRNA ANRIL has been indicated to protect cardiomyocytes from hypoxia and ischemia-induced loss through multiple signaling pathways. Shu et al. found that lncRNA ANRIL protected H9C2 cells from hypoxia-induced injury via miR-7-5p/SIRT1 axis [89], and Shi et al. demonstrated that LncRNA ANRIL regulated cardiomyocyte apoptosis through the IL-33/ST2 pathway [90]. According to reports, camk2d-related transcript 1 (C2dat1) is a lncRNA associated with various ischemic diseases [91]. Huan et al. discovered that C2dat1 may negatively regulate miR-22, up-regulated the expression of VEGF and enhanced the activation of PI3K/AKT/mTOR and JAK/STAT3 pathways to jointly protect cardiomyocytes [92]. LncRNA 2810403D21Rik/Mirf acts as a competitive endogenous RNA of miR-26a. Inhibiting lncRNA 2810403D21Rik/Mirf expression can promote upregulation of miR-26a and autophagy to reduce heart damage [93]. A cell experiments demonstrated that lncRNA TUG1-miR-124-Hic-5 mediated survival of H9C2 cells [94]. However, in view of Wu et al, overexpression of TUG1 aggravated hypoxia-induced cell injury by modulating the miR-145-5p-Binp3 axis and activating the Wnt/catenin signaling pathway in H9C2 cells [95]. Two different results indicated that TUG1 may be a potential target for the treatment of AMI, but the specific mechanisms deserved further studied. In AMI rats, H19 can be used as an endogenous sponge to competitively bind to miR-22-3p and up-regulate the expression of KDM3A, thereby protecting myocardium from AMI-induced damage [96]. Du et al. revealed that silencing the expression of GAS5 can reduce hypoxia-induced apoptosis of H9C2 cells, and protective mechanism may be that lncRNA GAS5 acted as miR-142-5p molecular sponge to inhibited the further expression of lncRNA GAS5 [97]. LncRNA MEG3 has also been found to increase FoxO1 activity under hypoxic conditions and promote cardiomyocyte apoptosis [98]. It is reported that suppressing the expression of lncRNA XIST can inhibit myocardial cell apoptosis in AMI rats by down-regulating miR-449 and up-regulating miR-130a-3p levels [99,100]. Both in cell and animal experiments, LncRNA MALAT1 was verified to play an important role in myocardial ischemia through various signaling pathways, including inhibiting the ERK/MAPK pathway, regulating Sirt1 mediated by miRNA-217 and downstream PI3K/AKT as well as Notch signaling pathway [101,102]. HOX antisense intergenic RNA (HOTAIR), a 2.2 kb lncRNA, was originally described as a modulator of HOX gene expression. A recent study observed that adenoviral vector-driven HOTAIR overexpression significantly inhibited hypoxia-induced cardiomyocyte apoptosis partially based on the negative regulation of miR-1, while the adenovirus short hairpin HOTAIR knockout showed the opposite result [103]. Subsequently Zhang et al. discovered that HOTAIR also could be a sponge of miR-519d-3p in animal models. HOTAIR together with miR-519d-3p can alleviate myocardial infarction by inhibiting cardiomyocyte apoptosis and necrosis [104]. In oxygen-glucose deprivation (OGD) induced cardiomyocyte injury experiments, silencing circ_0010729 expression can activate mTOR and MEK/ERK pathways by up-regulating miR-145-5p, thereby protects cardiomyocytes from OGD-induced damage [105]. Antisense to the cerebellar degeneration related protein transcript, Cdrlas, also known as circular RNA sponge for miR-7, ciRS-7, recently and study showed that Cdrlas and miR-7a increased in a time-dependent manner within 24h after MI or 12 h after hypoxia treatment, which suggested that Cdrlas and miR-7a may play important roles in MI-induced cardiomyocyte apoptosis. The anti-apoptotic mechanism may be that Cdrlas by inhibiting the function of miR-7a, upregulates the expression of target genes PARP and SP1, aggravates hypoxia-induced myocardial cell apoptosis and promotes myocardial infarction in mice [106] (Table 3).
NcRNAs in angiogenesis in MI region
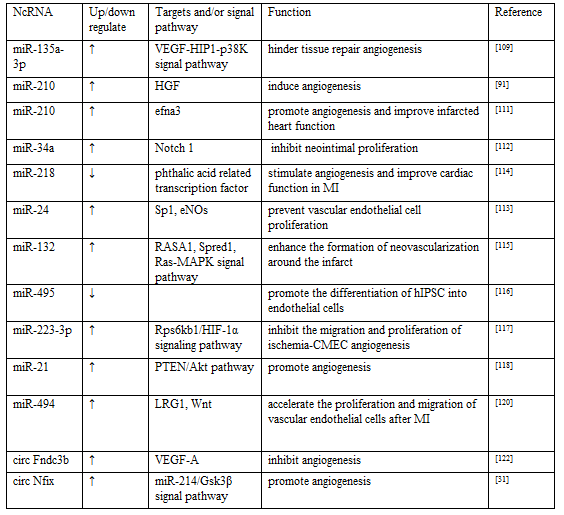
Angiogenesis is an important part of cardiac repair after MI. It generates new blood vessels to restore blood flow and remove necrotic cell debris, promote tissue perfusion recovery, and transport nutrients and cytokines, which are of benefit to myocardial tissue repair after MI. It is reported that the degree of angiogenesis is related to myocardial remodeling following MI, and has an important effect on the prognosis of patients [107]. VEGF is one of major signaling molecule for angiogenesis. In recent years, ncRNAs have been found to promote or hinder angiogenesis after MI by regulating VEGF expression levels and multiplying downstream signaling pathways [108]. it is reported that miR-135a-3p hindered tissue repair angiogenesis by targeting the VEGF-HIP1-p38K signaling axis [109]. MiRNA-210 has been reported to have similar angiogenic effects [110]. miR-210 is a key regulator of angiogenesis and endothelial cell survival under hypoxia [91], However there is still some disputes about its specific regulatory molecular mechanism. Fan et al. claimed that miR-210 induced angiogenesis by up-regulating hepatocyte growth factor (HGF) expression rather than act in the VEGF target gene. HGF is similar to VEGF and is an important stimulator of angiogenesis [91]. Na et al. proposed that miR-210-efna3 dependent mechanism involved in process of angiogenesis. They found that miR-210 was highly enriched in MSC-extracellular vesicle (EVs), which could reduce the expression of Efna3 in human umbilical vein endothelial cells, then promote angiogenesis and improve infarcted heart function, conversely the angiogenesis effect of miR-210 silenced MSC-EVs disappeared [111]. These studies indicate that miRNAs can regulate vascular activity after MI through different regulatory factors and signaling pathways.
Notch homologue 1, which was predicted as one of the primary targets of miR-34a, is a regulator of vascular smooth muscle cell (VSMC) function and arterial reconstruction. miR-34a inhibited VSMC and neointimal proliferation by regulating the expression level of Notch gene [112]. Zhang et al. reported that miR-24 prevented endothelial nitric oxide synthase expression and vascular endothelial cell proliferation by regulating Sp1 [113, while the down-regulation of miR-218 could inhibit phthalic acid related transcription factor to stimulate angiogenesis and improve cardiac function in MI rats [114]. Ma et al. found that injection of MSC-derived exosomes rich in miR-132 into ischemic myocardium of rats significantly enhanced the formation of neovascularization around the infarct. The molecular mechanism is that miR-132 directly inhibits RASA1 and Spred1 to enhance the Ras-MAPK signaling pathway [115]. According to the analysis of miR-spectrum and bioinformatic, miR-495 was thought to have anti-angiogenesis potential. LIANG et al. discovered that inhibition of miR-495 could promote the differentiation of human induced pluripotent stem cell (hIPSC) into endothelial cells, enhance therapeutic angiogenesis of hIPSC around the infarct edge [116]. MiR-223-3p has been identified as the core miRNA of ischemia- cerebral microvascular endothelial cells (CMEC) angiogenesis, which can inhibit the migration and proliferation of ischemia-CMEC angiogenesis by regulating the Rps6kb1/HIF-1α signaling pathway, thereby weakening the ability of angiogenesis [117]. Recently, in pre-clinical animal models of MI, it was found that EVs equipped with miR-21 effectively introduced miR-21 into cardiomyocytes and endothelial cells, then activated PTEN/Akt signaling pathway, stimulated endothelial cells to express VEGF and promoted angiogenesis [118]. The above studies indicated another potential role of miR-21 in regulating angiogenesis in addition to regulating myocardial fibrosis. Similar to miR-21, miR-27b also has a protective effect on the angiogenesis and infarcted heart function [119]. Recently, as Su et al. reported that leucine-rich alpha-2-glycoprotein-1 (LRG1) is negative regulated by miR-494, further found that over-expression of miR-494 may inhibited LRG1 and inactivated the Wnt signaling, thus accelerating the proliferation and migration of vascular endothelial cells after MI [120]. In recent years, although many miRNAs have been found to play roles in the mediation of vascular activity after MI, however, most of these studies are superficial and isolated, and the miRNA with good effects and it's deeper mechanisms still need to be further explored and discovered.
Even though some studies have shown that some lncRNA and circRNA are relevant to the regulation of the behavior of vascular endothelial cells and VSMC in a large number of tumors and cerebrovascular disease models [121], however there are a few studies which revealed the specific regulation of the angiogenesis of lncRNA and circRNA after MI in recent years. Garikipati et al. pointed out the view that a high level of circFndc3b in the ischemic myocardium can inhibit angiogenesis, the main mechanism was that circFndc3b enhanced the expression and signal transduction of VEGF-A [122]. CircNfix has been found that not only adjusts the potential of cardiac muscle cell proliferation, but also can plays an important role of angiogenesis regulation via circNfix/miR-214/Gsk3β pathway after MI [31]. Although there are few studies on role of lncRNA and circRNA in angiogenesis after MI, their ability to modulate the angiogenic activity of tumor cells suggests that they may also have a certain therapeutic potential in modulating the vascular activity following MI, which needs to be actively explored by more scientists (Table 4).
In summary, ncRNAs, such as miRNA, lncRNA and circRNA can individually or synergistically regulate the repair process after MI, which involved in the process of cardiomyocyte apoptosis, myocardial regeneration, fibroblast activation and angiogenesis following MI. MI is a complex process with multiple stages and successive activation of various cytokines and mechanisms. ncRNAs have multiple targets and can form circRNA/ lncRNA-miRNA-mRNA regulatory network, which all suggest that ncRNAs are potential therapeutic targets for repair of MI and is worthy of further in-depth study.
Competing interest: None declared.
Funding: This research was funded by NFSC (81860783)
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
