
Case Report | DOI: https://doi.org/10.31579/2642-973X/005
1 Molecular Biology & Genetics Harvard University. Director of the Division of Medical Genetics and Molecular Research. Division of Medical Genetics and Molecular Pathology Research. Massachusetts, Harvard University of Medical Sciences.
*Corresponding Author: hahin Asadi –Molecular Biology & Genetics Harvard University. Director of the Division of Medical Genetics and Molecular Research. Division of Medical Genetics and Molecular Pathology Research. Massachusetts, Harvard University of Medical Sciences, Depar
Citation: Shahin Asadi. (2019). The role of genetic mutations in genes HSPB1 & HSPB8 in Distal Hereditary Motor Neuropathy, type II Syndrome. Brain and Neurological Disorders. 2(1); DOI: 10.31579/2642-973X/005
Copyright: © 2019, Shahin Asadi, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 04 February 2019 | Accepted: 04 March 2019 | Published: 04 March 2019
Keywords: DHMN2 syndrome,HSPB1, HSPB8 genes, Motor Neuropathy, Brain disorder
Symptoms of DHMN2 usually begin from the age of 10 to the middle of adulthood. Early signs of this disorder include clogging or weakness in the muscles of the toe and the next, the whole leg. The DHMN2 syndrome is caused by the mutation of the HSPB1 gene, which is positioned in the long arm of chromosome 7 as 7q11.23, and the HSPB8 gene, which is based on the long arm of chromosome 12, is 12q24.23.
Generalizations of Distal Hereditary Motor Neuropathy, type II Syndrome (DHMN2)
DHMN2 syndrome is a progressive genetic disorder that affects neurons in the spinal cord. This syndrome causes muscle weakness and affects movement, especially in the legs.
Clinical Signs and Symptoms of Distal Hereditary Motor Neuropathy, type II Syndrome (DHMN2)
Symptoms of DHMN2 usually begin from the age of 10 to the middle of adulthood. Early signs of this disorder include clogging or weakness in the muscles of the toe and the next, the whole leg. Over a period of about 5 to 10 years, people with DHMN2 syndrome experience a gradual decrease in muscle tissue (atrophy) in the lower legs. They are barely able to walk and run, and eventually they may have full paralysis of the legs. Thigh muscles may also be affected, although this usually happens late and less severe.

Figure 1: Images of skeletal disorders associated with DHMN2 syndrome.
Some people with DHMN2 syndrome undermine the muscles of the hands and the forearms. This weakness is less common in the lower limbs and is not usually caused by paralysis.

Figure 2: A child's foot image of DHMN 2 syndrome with foot disorder.
Etiology of Distal Hereditary Motor Neuropathy, type II Syndrome (DHMN2)
The DHMN2 syndrome is caused by the mutation of the HSPB1 gene, which is positioned in the long arm of chromosome 7 as 7q11.23, and the HSPB8 gene, which is based on the long arm of chromosome 12, is 12q24.23. These genes provide instructions for protein synthesis called the thermal shock beta protein 1 and the beta-8 thermal shock protein. Thermal shock proteins help protect cells in adverse conditions such as infection, inflammation, exposure to toxins, high temperatures, damage, and disease. They block the signals that lead to the death of the planned cells. Additionally, they appear to be involved in activities such as cell movement (mobility), structural cellular structure stability (cellular skeleton), The secretion and stabilization of freshly produced protein and the repair of damaged proteins are involved. Thermal shock proteins appear to play an important role in stimulating muscle fibers (muscle contraction).

Figure 3: Schematic view of chromosome 7, whose HSPB1 gene is located in the long arm of this chromosome 7q11.23.
The thermal shock beta-1 protein and the β-8 thermal shock protein are found in the body cells and are abundant in neural cells. In neural cells, the β-1 thermal shock protein helps to protect a network of molecular strands called neurofilaments with special bandwidths called axons. Proper maintenance of axon diameter is essential for the effective transfer of neural pathologies. The Beta-8 thermal shock protein function is not well understood, but studies have shown that it affects the axons with a thermal shock beta-1 protein.
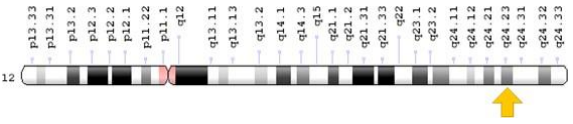
Figure 4: Schematic view of chromosome 12, in which the HSPB8 gene is located in the long arm of this chromosome 12q24.23.
HSPB1 and HSPB8 mutations that cause DHMN2 syndrome alter single-protein building blocks (amino acids) in protein sequences. If the protein changes, they may most likely close together and form aggregates. Thermal shock protein compounds may block the transport of substances essential for the proper functioning of neuronal axons. Disrupting the function of other cells that these proteins are involved in may also contribute to the symptoms and symptoms of DHMN2 syndrome.

Figure 5: Radiological Images of the Brain of People with DHMN2 Syndrome.
DHMN2 syndrome follows the dominant autosomal inheritance pattern. Therefore, to produce this syndrome, a copy of the mutated genes of HSPB1 and HSPB8 (parent or parent) is needed and the chance of having a child with this syndrome in the dominant autosomal state is 50% for each pregnancy.
Frequency of Distal Hereditary Motor Neuropathy, type II Syndrome (DHMN2)
DHMN2 syndrome is a hereditary genetic disorder whose frequency is not known in the world. At least 25 cases of this syndrome have been reported in medical literature from around the world.

Figure 6: A schematic view of the dominant autosomal inheritance pattern that follows DHMN2 syndrome.
Diagnosis of Distal Hereditary Motor Neuropathy, type II Syndrome (DHMN2)
The DHMN2 syndrome is diagnosed based on the clinical and physical findings of the patients and some pathological tests. The most accurate method for detecting this syndrome is the molecular genetic testing of HSPB1 and HSPB8 genes to investigate the presence of possible mutations.
Therapeutic routes for Distal Hereditary Motor Neuropathy, type II Syndrome (DHMN2)
The treatment and management strategy for DHMN2 syndrome is symptomatic and supportive. Treatment may be done by a team of experts, including orthopedic surgeons, neurologists, physicians, and other health care professionals. There is no definite treatment for this syndrome and all clinical measures are needed to reduce the suffering of the infected person. Genetic counseling is also important for all parents who want a healthy baby.
DHMN2 syndrome is a progressive genetic disorder that affects neurons in the spinal cord. This syndrome causes muscle weakness and affects movement, especially in the legs. Symptoms of DHMN2 usually begin from the age of 10 to the middle of adulthood. Early signs of this disorder include clogging or weakness in the muscles of the toe and the next, the whole leg. The DHMN2 syndrome is caused by the mutation of the HSPB1 gene, which is positioned in the long arm of chromosome 7 as 7q11.23, and the HSPB8 gene, which is based on the long arm of chromosome 12, is 12q24.23. There is no definite treatment for this syndrome and all clinical measures are needed to reduce the suffering of the infected person.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
