
Case Report | DOI: https://doi.org/10.31579/2690-4861/040
*Corresponding Author: Anubha Bajaj, Consultant Histopathologist, A.B. Diagnostics, A-1, Ring Road, Rajouri Garden, New Delhi, India.
Citation: Anubha Bajaj. (2020) The Paraneoplastic Presence- Phosphaturic Mesenchymal Tumour. International Journal of Clinical Case Reports and Reviews. 3(4); DOI: 10.31579/2690-4861/040
Copyright: © This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 10 July 2020 | Accepted: 27 August 2020 | Published: 09 September 2020
Keywords: Key words
Abstract
Phosphaturic mesenchymal tumour is a unique neoplasm of bone and soft tissue delineating diverse, specific histological features along with production of FGF-23 and associated metabolic bone defects such as rickets and osteomalacia. Paraneoplastic syndrome engendered by the neoplasm is associated with an acquired variant of oncogenic osteomalacia (OO), which can also appear in diverse mesenchymal neoplasia. Reoccurring tumefaction with oncogenic osteomalacia (OO) can emerge within sites such as mandible along with pulmonary and soft tissue metastasis.
Although the neoplasm can delineate intermediate malignant potential and unpredictable biological behaviour, distant metastasis is exceptional. Phosphaturic mesenchymal tumour was initially described by Mc Cane in 1947 with subsequent concordance between the neoplasm and osteomalacia [1]. Subsequently, Weidner and Cruz denominated the terminology of “phosphaturic mesenchymal tumour” (PMT) in 1987, adequately describing the morphological characteristics [2]. Although osteomalacia is manifest, phosphaturic mesenchymal tumour can be histologically misinterpreted. Occurrence of phosphaturic mesenchymal tumour in young individuals with skeletal deformities requires a distinction from disorders such as rickets or vitamin D deficiency. Elderly individuals depicting incrimination of ribs or multiple vertebral bodies may be indicative of multiple myeloma.
Disease Characteristics
Although phosphaturic mesenchymal tumour can arise anywhere within the bone or soft tissue, appendicular skeleton and extremities (95%) are commonly implicated whereas head and neck (5%) lesions are exceptional. Within the head and neck, tumour localization is discerned within sino nasal cavity (57%) followed by mandible (20%) [3].Also, lesions can emerge within chest wall, pharynx, maxilla, tongue, breast, spine, floor of mouth and posterior neck. Extra oral tumefaction is frequent in females with a female to male proportion of 2:1. Intraoral tumours delineate a mild male predominance. Tumours arising within hard and soft tissue are equivalently distributed with a slight predilection of bony sites, such as distal radius [3, 4].Median age of disease representation is 45 years whereas the condition is commonly discerned between 24 years to 58 years, although young subjects can be incriminated and no age of disease occurrence is exempt. A slight female predilection is observed with female to male ratio of ≈ 1.7:1. Specific factors of probable disease emergence are absent. Tumefaction is miniature and gradually progressive, thus several or a mean of 5 years is necessitated for adequate tumour localization.Incidence of phosphaturic mesenchymal tumour arising within head and neck may be enhanced as the neoplasm is frequently misinterpreted as glomangiopericytoma, giant cell tumour or low grade osteosarcoma. Phosphaturic mesenchymal tumour comprises of majority (80%) of mesenchymal neoplasms engendering oncogenic osteomalacia (OO) whereas around 20% of osteomalacia associated neoplasms are adjunctive mesenchymal tumefaction such as haemangiopericytoma, giant cell tumour of bone, osteosarcoma, fibrous dysplasia, aneurysmal bone cyst or chondromyxoid fibroma[3,4].Phosphaturic mesenchymal tumour produces fibroblast growth factor -2 (FGF-2), a protein which inhibits reabsorption of phosphate or dentin matrix protein1 by renal tubules [3].An estimated 60% neoplasms arising within non head and neck sites can harbour a chromosomal translocation with production of contemporary FN1-FGFR1 fusion protein, which engenders an upregulation of FGFR1 receptor[3,4].Majority of phosphaturic mesenchymal tumours are benign whereas an estimated 10% neoplasms relapse. Malignant metamorphosis is accompanied with multifocal or metastatic disease.Multiple, miniature, non-obstructive foci of pulmonary tumour metastasis can be discerned following repetitive surgeries. Malignant neoplasms can metastasize to nasal cavity, lip or tongue [3, 4].
Disease Pathogenesis
The neoplasm frequently engenders tumour induced osteomalacia (TIO) which is a paraneoplastic syndrome characteristically delineating phosphate wasting within renal tubules with subsequent hypophosphatemia [4].Osteomalacia is an adult metabolic disorder incorporating mineralization of mature bone and is generated by vitamin D deficiency, various medications, malabsorption, hepatic or renal disorders. Oncogenic osteomalacia (OO) is an exceptional clinico-pathological condition of acquired, secondary osteomalacia arising due to phosphaturic mesenchymal tumour [4, 5]. Phosphaturic mesenchymal tumour induces renal phosphate decimation with consequent osteomalacia via production and secretion of phosphatonins, commonly fibroblast growth factor 23 (FGF-23). Phosphatonins circumvent reabsorption of phosphate within proximal renal tubules with resultant hypophosphatemia and subsequent bone depletion. Overexpression of FGF-23 is accompanied by fibronectin-1-fibroblast growth factor -1 (FN1-FGFR1) fusion, as discerned by reverse transcriptase polymerase chain reaction (RT-PCR), a feature which is manifest in a subset of neoplasms. As FN1-FGFR1 fusion gene is discerned in around 60% of neoplasms, it is posited to be the pathway of tumouri-genesis [4, 5]. A subset of phosphaturic mesenchymal tumours arising within head and neck harbour FGFR1 chromosomal translocation, an anomaly which can be adequately discerned with fluorescent in situ hybridization (FISH), beneficially adopted in instances of obscure clinical and laboratory manifestations[5].Biochemical assay delineates enhanced bone turnover with hypophosphatemia, hyper-phosphaturia, elevated alkaline phosphatase, normal to minimal circulating levels of 1,25 dihydroxy-vitamin D3 (1,25-[OH]2D3), normal serum calcium and parathyroid hormone (PTH), findings which are consistent with oncogenic osteomalacia (OO) [5].Phosphate imbalance arises due to excessive expression of phosphatonin or fibroblast growth factor 23 (FGF-23), an ectopic, hormone-like protein secreted by phosphaturic mesenchymal tumour. Enhanced levels of FGF-23 engender phosphate diuresis or phosphaturia which prohibits reabsorption of renal phosphate with consequent hypophosphatemia. Aforesaid manoeuver activates the mobilization of phosphate and calcium from bone into circulating blood as a compensatory mechanism with weakening of bones and consequent fractures. Thus, enhanced expression of FGF-23 is contemplated to be diagnostic of phosphaturic mesenchymal tumour. As FGF-23 inhibits transportation and reabsorption of phosphate within proximal renal tubules, metabolism of vitamin D is influenced with circumvention of conversion of 25- hydroxy-vitamin D to 1,25- dihydroxy vitamin D(5,6).Although precise mechanism of FGF-23 influencing phosphate equilibrium is obscure, it’s function is separate from type IIa sodium phosphate co- transporter (NaPi-2a), a molecule which is regulated by parathyroid hormone (PTH) and significantly constitutes towards renal phosphate reabsorption. Additionally, heparin- like molecules and mitogen-activated protein kinase (MAPK) pathway may be pertinent for activity of FGF-23 [5, 6].
Clinical Features
The neoplasm is subdivided into four distinct categories as phosphaturic mesenchymal tumour
•mixed connective tissue type which occurs within soft tissue
•osteoblastoma- like
•ossifying fibroma –like
•non ossifying fibroma –like.
Aforesaid categories are discerned within diverse bony sites. Bone and soft tissue lesions appear as a singular entity or as components of a wide histological spectrum [5, 6].A subset of tumours may not demonstrate phosphate diuresis and are labelled as non phosphaturic variant of phosphaturic mesenchymal tumour.Clinical representation is contingent to anatomic distribution of lesions and is manifest by bone pain, multiple bone fractures, muscle tenderness, anomalies of gait, atrophy of proximal muscles and osteopenia [5, 6].Typically, pain and weakness is denominated. Numerous pathologic fractures within the spine, ribs, sacrum or calcaneus can appear. Destructive, osteolytic lesions can arise within incriminated bones. Adolescent subjects can depict rickets- like skeletal deformities.Symptoms of hypophosphatemia or osteomalacia can ensue with chronic pain or pathological fractures. Tumour discernment can be challenging in individuals demonstrating an absence of osteomalacia.Subjects can exhibit chronic lumbar pain with osteopenia of lumbar spine and neck of femur, osteoporosis of diverse bones as distal third of radius and history of atraumatic rib fractures [5, 6].Clinical symptoms are pertinent to implicated tumour site. Also, osteomalacia may not a comprehensive clinical feature. The neoplasm may not be clinically evident at representation and discernment is often delayed. Thus, incriminated subjects can be preliminarily treated for hypophosphatemia and osteoporosis [6].
On examination, dome shaped, symmetrical, non-ulcerated lesions of varying magnitude at intra-oral or diverse sites can be discerned. Phosphaturic mesenchymal tumour can be devoid of accompanying tumour induced osteomalacia (TIO) and manifest normal serum phosphate levels at initial disease representation. Tumour localization with early disease onset, prior to appearance of TIO is pertinent in aforesaid instances [5, 6].
Histological Elucidation
The mesenchymal, soft tissue or bony, variably cellular neoplasm with an infiltrative perimeter characteristically demonstrates foci of pale grey, flocculent or “grungy” calcification. Tumour magnitude varies from 2 centimetres to 14 centimetres.Infiltrative, hypo-cellular tumefaction is composed of spindle-shaped cells, irregular or miniature foci of “grungy” or flocculent calcification and an encompassing chondromyxoid or osteoid- like matrix with plump, fibroblastic cells [6].Characteristically, proliferating spindle- shaped cells are intermixed with several pseudo-vascular spaces and calcified tumour matrix. Well defined, spindle –shaped, stellate or epithelioid cells with intermixed multinucleated giant cells and circumscribing chondromyxoid matrix denominate the neoplasm. Focal micro- mineralization envelops individual tumour cell. Singular, cellular subtype can predominate in a specific neoplasm. Zones of flocculent or “grungy” calcification are interspersed within tumour cells [6, 7].Uniform, spindle- shaped cells are benign appearing and demonstrate miniature nuclei, well dispersed nuclear chromatin and indistinct nucleoli. Haemagiopericytoma-like vascular pattern, distinct foci of “grungy” calcified matrix, mature adipose tissue, focal microcyst formation and haemorrhage, an incomplete perimeter of membranous ossification and zones of metaplastic bone formation are discerned. Osteoid- like matrix or spindle-shaped cellular proliferation can occasionally display an absence of multinucleated giant cells or calcification. Thus, tumour discernment in aforesaid instances is challenging [6, 7].Infiltrative, storiform or fascicular tumour pattern can be observed. Mitotic activity is minimal to absent. Tumour is devoid of atypia.As the significantly vascular neoplasm is characteristically configured by uniform, spindle-shaped cells and numerous, disseminated multinucleated giant cells enveloped within a chondromyxoid matrix with focal calcification, stromal alterations may vary from myxoid to hyalinised to abundantly collagenous to zones of osteoid-like matrix, admixed punctate or flocculent calcification[6,7].Extensively cellular neoplasm exemplifies numerous, osteoclast –like, multinucleated giant cells. Neighbouring vascular articulations are thin walled and impart a sieve- like appearance to the tumefaction. Foci of red cell extravasation are observed [7].Enhanced tumour cellularity, hyperchromasia or significant cellular pleomorphism is discerned.Cellular neoplasm configured of spindle-shaped cells and osteoid- like matrix is designated as an “ossifying” subtype of phosphaturic mesenchymal tumour [6, 7].Extensive metastatic disease can ensue in advanced cases with mitotic activity ranging from 10 to 25 per 10 high power fields. Aggressive or metastatic tumefaction delineates a proliferative index Ki67 of around ≈ 3% [7].Malignant metamorphoses of phosphaturic mesenchymal tumour is exceptional and challenging to discern as tumour expanses may resemble undifferentiated pleomorphic sarcoma or malignant fibrous histiocytoma. Malignant conversion is exhibited by nuclear atypia, mitotic figures exceeding >5 per 10 high power fields and enhanced tumour cellularity. Cytological features of the neoplasm may not be predictive of malignant biological behaviour [6, 7].On cytogenetic analysis, an FGFR1 chromosomal translocation is identified in around 80% of tumour cells. Also, complex chromosomal translocations can occur in nearly 42% of cells [7].
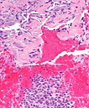
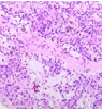
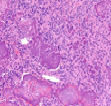
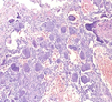
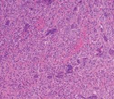
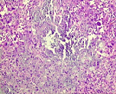


Immune Histochemical Elucidation
Phosphaturic mesenchymal tumour is immune reactive to vimentin, thereby indicating a mesenchymal origin. Additionally, the neoplasm is immune reactive to fibroblast growth factor 23 (FGF-23) and dentin matrix protein1 and is immune non-reactive to S100 protein, CD68, CD34, desmin or cytokeratin [3, 4].
Differential Diagnosis
Phosphaturic mesenchymal tumour requires a segregation from cementifying fibroma, myxoid neoplasm, benign chondromesenchymal tumour and giant cell tumour of soft tissue.Soft tissue neoplasms necessitating distinction are solitary fibrous tumour (SFT) and glomangiopericytoma, tumours which recapitulate the vascular configuration of phosphaturic mesenchymal tumour although lack chondromyxoid matrix and “grungy” or flocculent calcification[8].Bone tumours mandating demarcation are giant cell tumour, chondromyxoid fibroma, chondroma and chordoma[8].Mesenchymal neoplasms engendering oncogenic osteomalacia (OO) are haemangiopericytoma, giant cell tumour of bone and osteosarcoma.
• Giant cell tumour is characterized by proliferation of mononuclear and osteoclast- like giant cells, demonstrating identical nuclear features. Giant cell tumour of bone demonstrates numerous multinucleated giant cells composed of clear cytoplasm and an agglomeration of roughly 20 nuclei to 25 nuclei. Foci of spindle-shaped cells, typically configuring a storiform pattern and reactive, immature bone is delineated although chondroid matrix of phophaturic mesenchymal tumour is absent, which contains fewer multinucleated giant cells with fewer nuclei [8, 9].
• Chondromyxoid fibroma is a lobulated, well circumscribed lesion comprised of chondroblasts imbued with abundant, eosinophilic cytoplasm. Tumour cells are embedded within a myxoid or poorly configured, hyalinised, cartilaginous stroma. Periphery of tumour nodules delineated enhanced cellularity. Osteoclasts, zones of calcification and spindle-shaped cells are typically exhibited within fibrous septa. Although tumour vascularity is enhanced, blood vessels are disparate from haemangiopericytoma- like vascular configurations of phosphaturic mesenchymal tumour [8, 9].
• Chordoma is an exceptional, malignant bone tumour which arises from midline of foetal notochord. Dual cellular categories comprising the neoplasm are elliptical cells and physaliferous cells articulating cords and lobules, separated by fibrous tissue septa. Neoplastic cells are enmeshed within an extensively myxoid stroma with foci of calcification and accumulated multinucleated giant cells. However, predominant spindle-shaped cellular component is lacking. Chordoma is immune reactive to S100 protein, cytokeratin, epithelial membrane antigen (EMA) and brachury [8, 9].
• Cementifying fibroma is discerned within mandible, maxilla or adjunctive facial bones and is a well circumscribed neoplasm containing variable quantities of mineralized matrix simulating cementum. The neoplasm is composed of bland, spindle- shaped cells with foci of calcification, akin to phosphaturic mesenchymal neoplasm [8, 9].
• Haemangiopericytoma displays hyalinised, “staghorn” blood vessels layered by spherical to elliptical cells. Tumefaction lacks “grungy”, flocculent calcification and tumour associated multinucleated giant cells.
• Osteosarcoma histologically simulates phosphaturic mesenchymal tumour, engenders variable quantities of bone or neoplastic osteoid directly from tumour cells and displays cytological atypia. The neoplasm can manifest a significant cartilaginous component. In contrast, phosphaturic mesenchymal tumour is composed of bland, spindle-shaped cells although bone is also generated [8, 9].As osteosarcoma or chondrosarcoma require segregation, typical foci of phosphaturic mesenchymal tumour necessitate recognition and aid differentiation [8].
Preoperative biochemical analysis can exhibit hypo-phosphatemic syndrome concurrent with tumour induced osteomalacia (TIO) and consequent hyperphosphaturia.Enhanced expression of FGF-23 m RNA within neoplastic cells can be demonstrated by chromogenic in situ hybridization (CISH), efficaciously employed upon formalin fixed, paraffin embedded tissue. Molecular assay of serum FGF-23 can be beneficially adopted [8, 9].Contemporary FN1-FGFR1 fusion protein, which engenders an upregulation of FGFR1 receptor, can be discerned by reverse transcriptase polymerase chain reaction (RT-PCR), fluorescent in situ hybridization (FISH) or Western blot analysis [9].However, FGFR1 gene can demonstrate an alternative chromosomal translocation pattern which requires the adoption of multiple sequence specific probes, as discerned by reverse transcriptase polymerase chain reaction (RT-PCR). Break apart fluorescent in situ hybridization (FISH) assay can delineate chromosomal translocation of FGFR1 gene with associated partner genes [9].Subjects with TIO can manifest histological features concurrent with phosphaturic mesenchymal tumour and associated head and neck neoplasms such as glomangiopericytoma. Aforesaid tumours warrant adequate discernment with ancillary molecular techniques such as fluorescent in situ hybridization (FISH), which can confirm chromosomal translocation within FGFR1 gene or chromogenic in situ hybridization (CISH), in order to delineate elevated expression of FGF-23. In situ hybridization performed for FGF-23 demonstrates a diffuse, intense cytoplasmic staining. Aforesaid procedures can be performed on formalin fixed, paraffin embedded tissue [9].Occasionally, appropriate detection of miniature tumours can be challenging. Therefore, cogent methods of assessment such as magnetic resonance imaging (MRI), octreotide scintigraphy and positron emission computerized tomography (PET-CT) can be suitably adopted [9].Cogent factors in disease discernment are age and gender of incriminated individual, tumour localization, initial diagnosis, duration of symptoms, occurrence of tumour induced osteomalacia (TIO) with serum calcium and phosphate levels [9].
Subjects with extensive tumour induced osteomalacia (TIO) exceeding >10 years are initially managed with supplemental phosphate. Eventually, surgical resection of the neoplasm is curative and alleviates hypophosphatemia [8, 9].Adequate surgical eradication of the neoplasm with a broad perimeter of tumour-free tissue is a preferred mode of therapy. Elevated serum phosphate can occur within weeks following surgery, levels which can return to normal with pertinent surgical excision. Surgical eradication can suitably reverse metabolic effects engendered by the neoplasm.Tumour reoccurrence is absent. Also, clinical behaviour of the neoplasm is unpredictable and lacks concurrence with manifested cytological features. Thus, extended follow up is warranted [8, 9].Neoadjuvant chemotherapy can be concomitantly adopted with extensive surgical extermination, especially with metastatic disease. Adjunctive treatment strategies such as radiotherapy and chemotherapy can be contemplated for treating tumefaction unamenable to surgery [8, 9].
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
