
Research Article | DOI: https://doi.org/10.31579/2766-2314/097
1 MMK and SDM Mahila Maha Vidyalaya, Mysore, Karnataka, India.
2 Yuvaraja's college, Mysore, Karnataka, India.
*Corresponding Author: Sudha B S, Yuvaraja's college, Mysore, Karnataka, India.
Citation: Pavithra V., Sudha B. S (2023), The Molecular Docking Study of Interaction of Newly Synthesised Benzamide Appended by Pyrazolone Derivatives as Ligand Molecule with The Target Protien 6lu7 of Novel Corona Virus, J, Biotechnology and Bioprocessing, 4(2); DOI:10.31579/2766-2314/097
Copyright: © 2023, Sudha B S. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 31 March 2023 | Accepted: 10 April 2023 | Published: 20 April 2023
Keywords: acute kidney injury; canine; crrt; kim-1; prognosis
Several survivability predictors have been identified in acute kidney injury (AKI) affected humans managed with continuous renal replacement therapy (CRRT). The aim of this study was to evaluate various blood and urine parameters as survivability predictors in AKI affected dogs managed with CRRT. Twenty dogs that presented with AKI to a veterinary hospital were managed with CRRT. Physical examination, urinalysis, arterial/venous blood gas analysis, and ELISA for analysis of Kidney Injury Molecule-1 (KIM-1) and Cystatin-C (Cys-C) were performed on the first day of presentation (Pre-CRRT).
Survivability evaluated on day 21 post-CRRT indicated 12 survivors and 8 non-survivors. Pre-CRRT parameters were compared between survivors and non-survivors using independent t-tests. Of the various parameters analyzed, KIM-1 concentrations and anion gap (AG) were significantly different between survivors and non-survivors. Other parameters such as APACHE III score, Cys-C, renal failure index (RFI), and fractional excretion of sodium (FENa) were not significantly different between survivors and non- survivors. Results of this study suggest that KIM-1 and AG could be employed as survivability predictors in AKI affected dogs managed with CRRT.
Plants and bioactive compounds have played an important role in the development of several clinically useful therapeutic agents since time immemorial. The global health emergency of novel COVID-19 is due to severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). As if now, there are no approved drugs for the treatment of corona viral disease (COVID-19), although some of the drugs have been tried. The virtual interaction of the COVID-19 main protease in complex with the inhibitor N3 (Research Collaboratory for Structural Bioinformatics Protein Data Bank [PDB] ID: 6LU7), Hence this is chosen as target protein molecule.
The molecular docking approaches are used to model the interaction between a small molecule and a protein at the atomic level, which allow us to characterize the behavior of small molecules in the binding site of target proteins as well as to elucidate fundamental biochemical processes and study of its features. The docking process involves two basic steps they are prediction of the ligand conformation as well as its position and orientation within these sites (usually referred to as pose) and also assessment of the binding affinity.
Pyrazolones are proven to be valuable over wide range of applications in the pharmaceutical industries. The most classic approach to prepare pyrazolones is via the reaction between a β-ketoester, β- cyanoester or β-unsaturated esters and hydrazine derivatives of alkanes, arenes and heterocycles.
Synthesis of benzamide appended heterocycles was initiated, to expand the application of synthesized compounds as the useful drugs.
Plan for Synthesis of Ligands
Synthesis of substituted benzamides 3a-f was achieved by reacting aminophenol (1) and benzyol chloride (2) in presence of trimethylamine at room temperature for 24hrs, further synthesis of compounds 4a-f is accomplished by reaction of substituted benzamides 3a-f with ethyl bromoacetate in presence of potassium carbonate. The obtained acetates 4a-f were reacted with 80% hydrazine hydrate to yield substituted hydrazides 5a-f. These phenyl hydrazides 5a-f were used further for cyclisation with ethyl acetoacetate in presence of 20% tetrabutylammonium hydroxide in methanol with ethylene glycol as solvent to achieve substituted N-(4-(2-(5-methyl-3-oxo-2,3-dihydropyrazolyl-1-)2- oxoethoxy) phenyl) benzamide derivatives 6a-f which is our ligand molecule. The structure of synthesized ligands was confirmed by characterization using spectroscopic techniques.
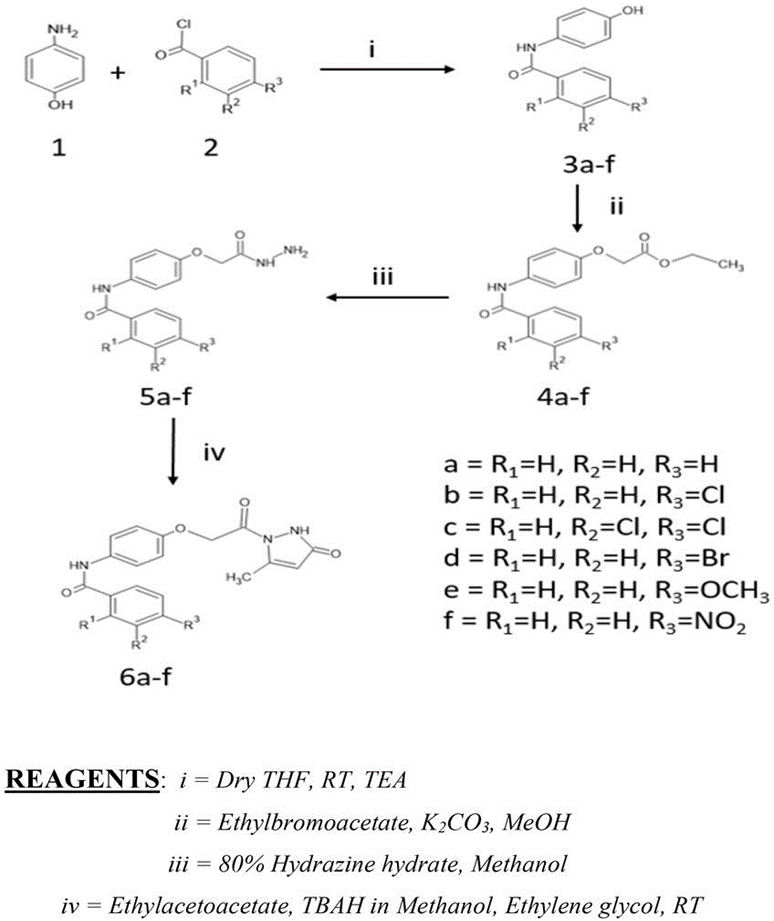
Figure 1: Scheme for Synthesis of Ligand Molecules
Chemicals and solvents used were purchased from Sigma Aldrich and used without further purification unless stated. Melting points were determined in open capillaries on a Buchi oil melting point apparatus and are uncorrected. Reactions were monitored by using thin layer chromatography (TLC) on aluminum sheet precoated with silica gel 60 F254 (0.2mm Merck). Chromatographic spots were visualized by UV light and /or with iodine. For column chromatography, silica gel of 100–200 mesh size was used. 1H NMR spectra were acquired on a Brucker NRC-IISC instrument, Banglore at ambient temperature at 400 and /or 300 MHz in DMSO-d6 or CDCL3 and TMS was used as an internal reference. 13C NMR spectra were recorded on a Bruker AMX-400(100 MHz) with complete proton decoupling. Chemical shift is reported in ppm from tetramethyl silane (TMS) with the solvent as the internal refence (CDCl3: δ 77.0 ppm or DMSO δ 40.0 ppm). LC-MS was performed on Agilent LCMS system equipped with a BEH C8, 30x4.6mm, 1.7µm column at a flow rate of 1.0ml/min.
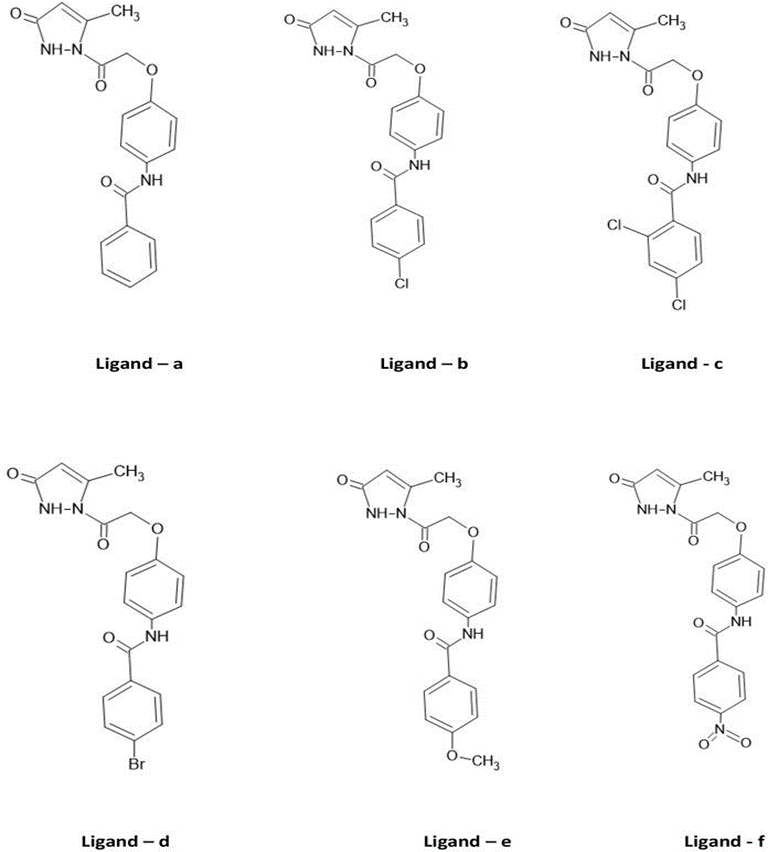
Figure 2: Structure of benzamide appended by pyrazlones acting as ligands
Molecular Docking
Molecular docking is a well-established computational technique which predicts the interaction energy between two molecules. This technique is basically incorporating the algorithms like ss dynamics, fragment search methods and soon. molecular docking also gives the clear idea that what biochemical mechanism is going on between protein and the ligand. In this article we are used to study the interaction of two molecules to fit the best orientation of ligand which would form a complex with overall minimum binding energy. We start with the in-silico generation of ligand and conversion of file format. Further selection of protein and preparation of protein by optimization of protein and energy minimization. Next using Autodock Vina 4.2 autodock was run to check the interaction and finally analysed by visualization tools and creation of algorithm table.

Table 1: Comparative study of interaction of ligand molecule with target protein 6LU7
Using commercial ACD/chemsketch tool we have drawn the ligand structure and saved in MDL mol. files and further this file is converted into PDB file format by using open babel 2.4.1 which is a necessary format to run autodock. The selected protein is downloaded from RCSB Protein Data Bank in PDB format. In this article we have selected COVID-19 main protease in complex with the inhibitor N3 (Research Collaborators for Structural Bioinformatics Protein Data Bank [PDB] ID: 6LU7. Next the protein optimization and energy minimization are done using swiss PDB viewer 4.1.0. by deleting all hetero atoms and co-ordinates.

Figure 3: Molecular interaction of ligands with molecules
Both ligand and protein molecules are prepared in PDB format which is necessary to run autodock. During docking all water molecules are removed from the protein and polar hydrogen atoms and kollman charges were added to the protein and assigned with AD4 types of atoms. Grid selected around 0.512 value was fixed for every run around the active site of protein that is GLN189 which is obtained by literature survey. The docked files are saved in DLG format by Lamarchian Genetic Algorithm and further it is used for visualization and tabulation of results.
The results of molecular docking and the poses of ligand- protein interaction was studied using autock tools for all interactions such as electrostatic forces, hydrogen bonding, torsional effects and so on.
Further visualization of the poses of ligand- protein interaction by Pymol. Specially to study the hydrogen bonding interaction of ligand and protein in the selected grid region. The interacted amino acids were tabulated and studied for further.
More the hydrogen bonds more will be the stability of the complex thus all six ligands were studied for their hydrogen bonding interaction with the same protein 6LU7. The selection of grid box size is same for all type of ligands used in the molecular docking studies. Totally ten runs were considered for the best pose of ligand-protein interaction among them the lowest binding energy is considered as the best pose. Further the selected interaction pose of ligand and protein were studied under pymol to get the hydrogen bonding interactions.
Interaction of ligand-a(N-{4-[2-(5-methyl-3-oxo-2,3-dihydro-1H-pyrazol-1-yl)-2- oxoethoxy] phenyl} benzamide)
After docking the protein target with each of our ligands, we have observed several information regarding the interactions. Each ligand has slight difference in their interaction type with the selected target protein 6LU7 molecule. According to it for ligand-a and protein 6LU7 interaction we found the least binding energy as -8.90 in the 8th run with the inhibition constant of 301.49 nM and the entropy of the selected grid was found to be 0.41. when the complex in viewed in pymol we found that Ligand-a is having three hydrogen bonds with the target protein molecule 6LU7 with the selected grid box. The bond length of the three hydrogen bonds is 2.0, 2.4 and 2.5 with the amino acids GLU166, SER144 and LEU141 respectively.
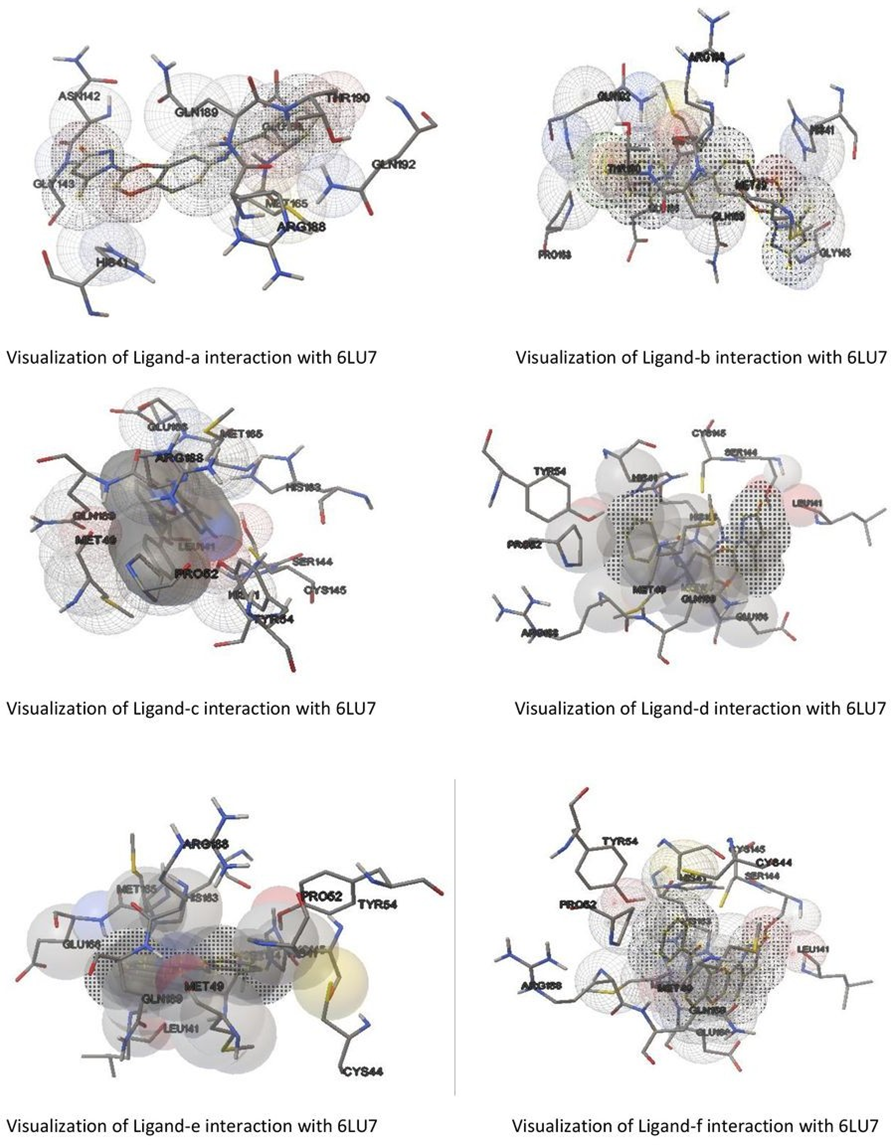
Figure 4: Visualization of protein-ligand interaction in terms of hydrogen bonding
Interaction of ligand-b (4-chloro-N-{4-[2-(5-methyl-3-oxo-2,3-dihydro-1H-pyrazol-1-yl)-2- oxoethoxy]phenyl}benzamide)
ligand-b and protein 6LU7 interaction we found the least binding energy as -8.77 in the 7th run with the inhibition constant of 374.22 nM and the entropy of the selected grid was found to be 0.11. when the complex in viewed in pymol we found that Ligand-a is having one hydrogen bonds with the target protein molecule 6LU7 with the selected grid box. The bond length of that one hydrogen bonds is 2.0 with the amino acids GLY143.
Interaction of ligand-c (2,4-dichloro-N-{4-[2-(5-methyl-3-oxo-2,3-dihydro-1H-pyrazol-1-yl)-2- oxoethoxy]phenyl}benzamide)
ligand-c and protein 6LU7 interaction we found the least binding energy as -8.99 in the 3rd run with the inhibition constant of 255.23 nM and the entropy of the selected grid was found to be 0.14. when the complex in viewed in pymol we found that Ligand-a is having one hydrogen bonds with the target protein molecule 6LU7 with the selected grid box. The bond length of that one hydrogen bonds is 2.9 with the amino acids ARG188.

Figure 5: Pymol viewer of hydrogen bondings in the protein-ligand complex
Interaction of ligand-d (4-bromo-N-{4-[2-(5-methyl-3-oxo-2,3-dihydro-1H-pyrazol-1-yl)-2- oxoethoxy]phenyl}benzamide)
ligand-d and protein 6LU7 interaction we found the least binding energy as -8.85 in the 7th run with the inhibition constant of 325.66 nM and the entropy of the selected grid was found to be 0.14. when the complex in viewed in pymol we found that Ligand-a is having one hydrogen bonds with the target protein molecule 6LU7 with the selected grid box. The bond length of that one hydrogen bonds is 3.2 with the amino acids GLU166.
Interaction of ligand-e (4-methoxy-N-{4-[2-(5-methyl-3-oxo-2,3-dihydro-1H-pyrazol-1-yl)-2- oxoethoxy]phenyl}benzamide)
ligand-e and protein 6LU7 interaction we found the least binding energy as -8.70 in the 3rd run with the inhibition constant of 420.33 nM and the entropy of the selected grid was found to be 0.10. when the complex in viewed in pymol we found that Ligand-a is having two hydrogen bonds with the target protein molecule 6LU7 with the selected grid box. The bond length of those two hydrogen bonds is 2.3 and 4.8 with the amino acids GLY143 and GLN189 respectively.
Interaction of ligand-f (N-{4-[2-(5-methyl-3-oxo-2,3-dihydro-1H-pyrazol-1-yl)-2-oxoethoxy]phenyl}-4- nitrobenzamide)
ligand-f and protein 6LU7 interaction we found the least binding energy as -9.17 in the 8th run with the inhibition constant of 190.41 nM and the entropy of the selected grid was found to be 0.15. when the complex in viewed in pymol we found that Ligand-a is having three hydrogen bonds with the target protein molecule 6LU7 with the selected grid box. The bond length of those three hydrogen bonds is 2.1, 2.3 and 2.1 with the amino acids GLN192, GLN192 and GLY143 respectively.
More the number of hydrogen bond in the complex more will be the stability and lesser the value of inhibition constant (Ki) more will be the interaction power of the ligand with protein which means a small concentration of ligand can show a good interaction with the protein molecule. When compare the interaction results of all the ligands with same selected protein target molecule, ligand-f is having a good interaction with protein when compare to all ligands because ligand-f is having binding energy as -9.17 in the 8th run with the inhibition constant of 190.41 Nm.
.
The data and visualisation images used in this research article are not plagiarised by anywhere.
Not applicable.
The whole article is written and data are obtained by author Smt. Pavithra V and the guidance for the research was provided by the corresponding author Dr. Sudha B S.
For this research article no funding is utilised and no funding agencies are involved.
The data base and software used for this research article are reproducible and available for universal usage.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
