
Case Report | DOI: https://doi.org/10.31579/2768-0487/121
1 Department of Laboratory Medicine, Shenzhen Second People’s Hospital, Shenzhen, China.
2 Clinical Laboratory, Department of Clinical Laboratory, The People’s Hospital of Longhua Shenzhen, Shenzhen, China.
*Corresponding Author: Dayong Gu, Department of Laboratory Medicine, Shenzhen Second People’s, Hospital Shenzhen, China.
Citation: Qiu C., Huang C., Chen X., Gu D., (2024), The Identification of a Novel Compound Heterozygous Mutation in Hereditary Human Coagulation Factor VII (FVII) Deficiency Following a Bamboo Leaf Green Snake Bite, Journal of Clinical and Laboratory Research. 7(3); DOI:10.31579/2768-0487/121
Copyright: © 2024, Dayong Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 13 November 2023 | Accepted: 15 January 2024 | Published: 28 February 2024
Keywords: hereditary factor VII (FVII) deficiency; venom-induced consumptive coagulopathy (VICC); gene mutation, pedigree analysis
Hereditary factor VII (FVII) deficiency is an uncommon autosomal recessive disorder associated with mutations in the F7 gene and laboratory investigations usually reveal isolated prolongation in prothrombin time (PT)/International normalized ratio (INR). Venom-induced consumptive coagulopathy (VICC) is distinguished by the activation of the coagulation pathway, which is triggered by procoagulant toxins in snake venom. Diagnosing snakebites in patients with hereditary FVII deficiency presents a challenge due to the fact that prolonged time PT/ INR is considered the most valuable diagnostic method for VICC. Therefore, it is possible that certain patients may not promptly receive an accurate diagnosis of hereditary FVII deficiency. We present a pedigree featuring hereditary FVII deficiency, which was diagnosed through Sanger sequencing, following a bamboo leaf green snake bite.
Coagulation FVII is a plasma vitamin K-dependent serine protease that is solely synthesized in the liver and its concentration in the bloodstream is approximately 0.5μg/ml. FVII(F7) gene, which is situated on chromosome 13q34, comprised a total of 9 exons [1]. FVII is a distinct coagulation factor that exhibits a minor portion (1perecentage–3perecentage) of its activated form (FVIIa) in the blood, even in the absence of coagulation activation [2]. A minute quantity of FVIIa can effectively initiate the process of coagulation. Coagulopathy is a prevalent outcome of snake bites, with venom-induced consumptive coagulopathy (VICC) being the most critical manifestation of this condition [3]. VICC is distinguished by the activation of the coagulation pathway, which is triggered by procoagulant toxins in snake venom. They are referred to as procoagulant toxins due to their ability to induce rapid clot formation in vitro and cause significant consumption of factors in vivo, leading to a potential risk of bleeding [4]. Hereditary FVII deficiency stands as the most prevalent among the rare autosomal recessive bleeding disorders, which is often referred to as a “chameleon disease” due to the absence of a clear association between plasma FVII levels and bleeding symptoms [5]. Diagnosing hereditary FVII deficiency becomes particularly challenging in cases of snakebite envenoming, due to the fact that the prothrombin time (PT)/International normalized ratio (INR) is considered the most valuable diagnostic method for VICC. Therefore, it is possible that certain patients may not promptly receive an accurate diagnosis of hereditary FVII deficiency. In this report, we present a pedigree featuring hereditary FVII deficiency in a 2-year-old boy who exhibits markedly low FVII activity and subsequently establish a diagnosis of the hereditary compound heterozygous FVII deficiency through Sanger sequencing, following a bamboo leaf green snake bite. The results revealed that the proband, who had severe FVII deficiency, identified compound heterozygosity for c. T1224G and c.G1034T mutations in the F7 gene, leading to p. His408Q and p. Gly345Val, respectively. One of these heterozygous variants c. G1034T: p. Gly 345Val was first reported.
A 2-year-old boy presented to the hospital with swelling and pain in his left foot, caused by a bite from a bamboo leaf green snake one day ago. Upon examination, there was noticeable bruising on the dorsal surface of the left foot without any signs of necrosis or fever present. The patient was promptly treated with antivenom neutralization of toxin, dexamethasone sodium phosphate for inflammation, azithromycin for infection, and furosemide for diuresis. During hospitalization, he exhibited normal health conditions with the exception of an abnormal coagulation profile, especially markedly prolonged PT/INR. Hence, it cannot be ruled out that the patient might have experienced concurrent venom-induced consumption coagulopathy (VICC). One month after being discharged, there was no longer any noticeable swelling and pain at the site of the snakebite. However, the value of PT/INR was still significantly higher than the upper reference range (Figure 1).
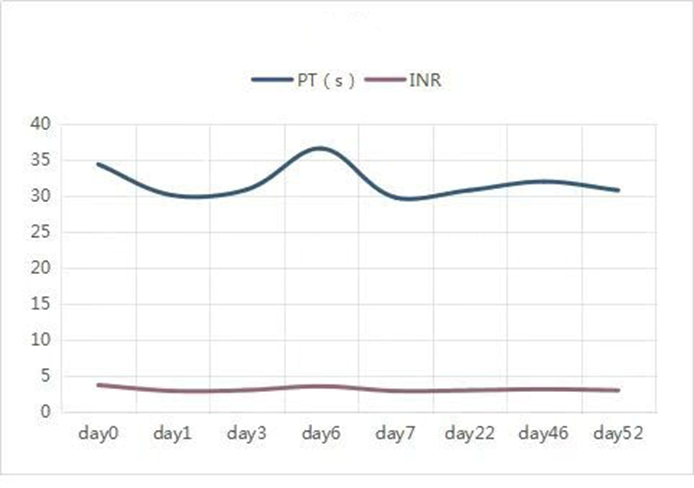
Figure 1: Demonstrates that the prothrombin levels in the proband did not return to normal until 52 days following the snake bite.
Further coagulation activities of the plasma FVII (FVII: C) assays confirmed that plasma FVII level was below 3Perecentage(normal range 70Perecentage-120Perecentage). Detection FVII: Ag by enzyme-linked immunosorbent assay (ELIAS) also showed a marked reduction. The mixing studies effectively restored the PT/INR to the normal range, suggesting a significant probability of a coagulation factor VII (FVII) deficiency. Meanwhile, his parents showed no abnormalities in the coagulation profile. The coagulation profile of the family is presented in Table 1.
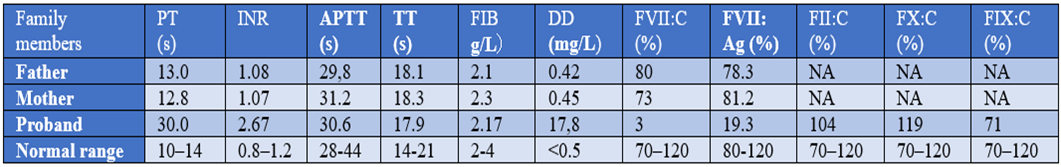
Abbreviations: INR, International standardized ratio of prothrombin; PT, prothrombin time; APTT, activated partial time; FIB fibrinogen; TT, thrombin time; DD, D – dimer.
Table 1: Coagulation profile in the family members.
Following the parent’s written informed consent, blood samples were collected for molecular testing and to establish an underlying genetic cause for coagulation factor VII (FVII) deficiency. Genomic DNA was extracted from the peripheral blood samples of all family members and all nine exons of FVII were analyzed using polymerase chain reaction (PCR) followed by next-generation sequencing at the Guangzhou Zhili Medical Laboratory. The PCR products that were sequenced and the corresponding GenBank sequences (NM_000131) were analyzed in order to identify the sites of mutation. The finding revealed that the proband exhibited a missense mutation (heterozygous) in exon 9 of the F 7 gene (c.T1224G, p. His 408 Q) was inherited from his mother and a missense mutation (heterozygous) in exon 9 of the F 7 gene (c. G1034T: p. Gly 345Val) was inherited from his father (Figure 2-3). No additional mutations were detected in the exons of the F7 gene. The proband was finally identified as a compound heterozygous carrier of the c.T1224G, p. His 408 Q and the c. G1034T: p. Gly 345Val mutations.
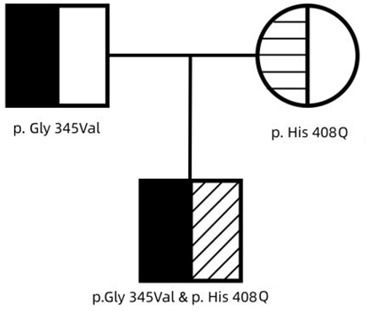
Figure 2: Pedigree of the family.
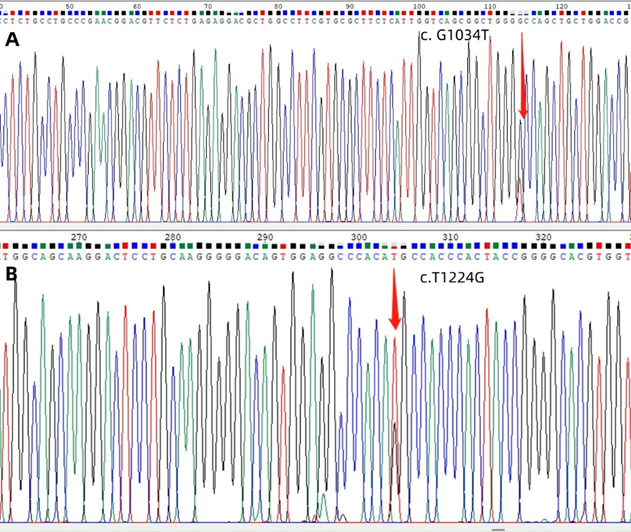
Figure 3: The Sanger sequencing revealed the presence of mutations in exon 9 of the F7 gene in pedigree. The mutation site c. G1034T was identified from the father, as shown in Panel A; Panel B displayed the mutation site c.T1224G from the mother.
Based on the reported data from http://www.factorvii.org [6], it is evident that the heterozygous mutation p. His 408 Q has already been reported, with a frequency of 0.0004 in East Asian populations. According to the American College of Medical Genetics and Genomics (ACMG) guidelines, the heterozygous mutation p. Gly 345Val has not been previously reported. All of the aforementioned mutations are disease-causing.
There are three conditions that can contribute to diagnosing FVII deficiency: laboratory tests conducted following bleeding episodes, screening within the scope of a family of FVII deficiency (when at least one family member carries a mutation), or incidental findings [7]. The median age at which the genetic defect was identified was 8 years [7]. FVII deficiency is readily suspected when the coagulation profiles reveal an isolated prolongation of PT and a normal activated partial thromboplastin time(aPTT), in conjunction with normal liver function [8]. To ensure the exclusion of transient or combined defects, it is recommended to draw samples a few weeks after the initial test and repeat the testing process for the confirmation of any congenital defects [9]. Extensive research studies conducted with clinical inquiries and encompassing extensive patient cohorts have significantly enhanced our comprehension of disease manifestation and diagnosis. Hereditary FVII deficiency presents a broad range of clinical manifestations, spanning from mild mucocutaneous bleeding (resembling platelet disorders) to potentially life-threatening bleeding, such as central nervous system (CNS) and gastrointestinal (GI) bleeding [10]. Approximately one-third of patients with deficiency typically remain asymptomatic throughout their lifetime [11]. The presentation of bleeding in FVII deficiency is highly variable, both in terms of location and severity. There is no direct correlation between FVII activity (FVIIa) and clinical findings [1]. In other words, the bleeding tendency in severely affected homozygotes or compound heterozygotes exhibits variability and does not consistently align with highly reduced FVII activity [10]. Therefore, the routine coagulation laboratory work-up, including PT and FVII levels based on PT is insufficient for actually assessing the risk of bleeding. This scenario was also observed in our patient, presenting significantly abnormal laboratory results, yet lacking abnormal clinical findings. The aforementioned results suggested that a minute quantity of FVIIa, even below 3perecentage, can effectively initiate the process of coagulation and protect from bleeding. The unpredictability of this behavior could possibly be attributed to the cumulative potentiation of the TF- FVII complex [12]. A complete absence of FVII has been observed to be correlated with life-threatening bleeding conditions in newborns [13]. However, a documented history of experiencing severe bleeding has been reported in patients with FVII: C levels above 5perecentage [14]. The reason for the absence of a correlation between clinical and laboratory phenotypes remains unclear. Whole gene sequencing (WES) is the preferred method for the genetic diagnosis of FVII deficiency. A small portion of the patients with FVII deficiency, less than 10%, do not identify any mutations using DNA sequencing. This could potentially be associated with a mutation in another gene [15]. Patients who shared the identical homozygous or compound heterozygous mutation did not consistently fall under the same severity classification, suggesting the existence of phenotypic diversity in the presence of the shared FVII mutations [16]. The structural analysis strongly reinforces the clinical findings, as it accurately predicts the presence of a structural defect that interferes with the function of the protein and causes a reduction in both FVII antigen and activity in plasma. We conducted a thorough structural prediction analysis of the patient’s mutated FVII protein. The green shape represents the normal protein, while the blue shape represents the mutated forms p. (Gly 345Val) and p. (His 408 Q) with valine and glutamine, respectively (Figure 4).
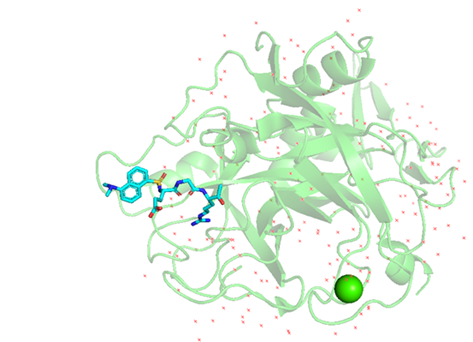
Figure 4: The figure displays the mutated protein derived from the PBD database with the purpose of identifying the structure of the factor VII (F7) protein. Especially, it represents the crystal structure of the active site-inhibited human coagulation factor VIIa (DES-GLA), available on RCSB PDB - 1CVW. Both mutations of the case were visualized on PyMOL. The normal protein was represented in green, while the mutated forms G1034T(Gly345Val) and T1224G (His 408Q) were displayed in blue. The mutated Valine and Glutamine were clearly visible in their respective positions.
Together with clinical presentation, laboratory work-up, and genetic findings, we report a novel compound heterozygous mutation, the heterozygous missense mutations of c.T1224G, p. His 408 Q and c. G1034T: p. Gly 345Val both in exon 9 of the F7 gene. These mutations potentially contribute to the congenital FVII defect in this family, suggesting a change in the molecular spatial conformation of the FVII domain.
We report a novel compound heterozygous mutation, the heterozygous missense mutations of c.T1224G, p. His 408 Q and c. G1034T: p. Gly 345Val both in exon 9 of the F7 gene. The identification of coagulopathy in a patient who has recovered from a snake bite is a significant diagnostic indicator for the presence of congenital coagulopathy.
There is no conflict of interest relevant to this paper to disclose.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
