
Case Report | DOI: https://doi.org/10.31579/2690-4861/033
*Corresponding Author: Anubha Bajaj, Consultant Histopathologist, A.B. Diagnostics, A-1, Ring Road, Rajouri Garden, New Delhi, India.
Citation: Anubha Bajaj. (2020) The Bairn’s Blain-Melanotic Neuroectodermal Tumour of Infancy. International Journal of Clinical Case Reports and Reviews. 3(2); DOI: 10.31579/2690-4861/033
Copyright: © 2020 Anubha Bajaj, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 10 July 2020 | Accepted: 07 August 2020 | Published: 11 August 2020
Keywords: Keywords
Abstract
Melanotic neuroectodermal tumour of infancy is an exceptional, benign, pigmented, melanin imbued neoplasm of neural crest origin, initially scripted by Krombecher in 1918[1]. The rapidly progressive neoplasm denominates a locally aggressive, biphasic tumefaction composed of miniature, primitive, round, neuroblast-like, blue cells admixed with enlarged, melanin- producing, epitheloid cells.The neoplasm is diversely nomenclated as melanotic progonoma, melanotic hamartoma, melanoameloblastoma, melanotic adamantinoma, congenital melanocarcinoma, retinal anlage tumour or pigmented congenital epulis Incriminated infants can depict elevated levels of urinary vanillylmandelic acid (VMA), thus implying a neural crest origin.
Disease Characteristics
Neural crest genesis of the neoplasm was hypothesized in 1966 on account of elevated urinary vanillylmandelic acid( VMA) secretion, as detected in adjunctive tumours of neural crest origin such as phaechromocytoma and neuroblastoma. Vanillylmandelic acid is restored to normal levels with surgical eradication of the neoplasm. Neural crest engenderment of the neoplasm can be determined by immune histochemical, ultrastructural and cell culture analysis. Also, biphasic cellular population comprised of melanocytic and primitive neuroectodermal cells appear as derivatives of neural crest [2, 3].On account of neural crest origin, gnathic bones are a frequent site of tumour incrimination. Majority (90%) of instances arise within cranio-facial sites and tumefaction is commonly discerned in maxilla (62.2%), skull (15%), mandible (7%), male reproductive system (5%), central nervous system (4%), soft tissues (1%) and female genital tract (<1%). Apart from head and neck, epididymis and testis are commonly incriminated besides exceptional implication of ovaries, soft tissues or bones of extremities [3]. Melanotic neuroectodermal tumour of infancy is typically discerned beneath < one year where majority (>90%) of incriminated subjects are infants. An estimated 50% of instances are beneath < 4.5 months. Median age of tumour discernment is 5 months. Congenital and prenatal cases are exemplified. A slight male predominance is observed.Although benign, melanotic neuroectodermal tumour of infancy can enhance rapidly and emerge as a locally infiltrative, destructive neoplasm with deformities ensuing in adjacent anatomic structures. Tumour reoccurrence is around 60%. Certain neoplasms undergo malignant metamorphoses wherein roughly 5% to 10% of tumefaction incur metastasis. Thus, preliminary tumour detection is mandated in order to minimize challenges and complications associated with treatment while inspiring a favourable outcome and normal developmental milestones [3, 4].
Characteristic genetic or molecular anomalies are absent in melanotic neuroectodermal tumour of infancy. The neoplasm is associated with BRAF-V600E mutation, germline heterogeneous mutation of CDKN2A with RPLP1-C19MC genetic fusion. Multiple tumour reoccurrences delineating a predominant neuroblastic component demonstrates loss of heterozygosity (LOH) with deletion of chromosome 1p and gain of chromosome 7q [3, 4].
Clinical Elucidation
Melanotic neuroectodermal tumour of infancy appears as a painless, expansive, lobulated, well circumscribed, un-encapsulated, firm, bluish- black, partially pigmented neoplasm with a superimposed intact epithelial surface. The tumefaction primarily arises within jaws of new born with frequent bone destruction and displacement of dental follicles. Appropriate discernment is contingent to pertinent clinical, radiographic and histological features although biphasic tumour composition is a crucial diagnostic element [3, 4].
Histological Elucidation
On gross examination, melanotic neuroectodermal tumour of infancy appears as greyish/ blue, firm, lobulated, well circumscribed tumefaction. Typically, tumefaction is superficially infiltrative and compresses abutting anatomic structures, instead of deep-seated tissue invasion. A blue- black hue is common on account of disseminated melanin. Cut surface is greyish to black contingent to quantity of melanin pigment [4, 5].Tumour nodules are comprised of neuroblast-like cells appearing as miniature, spherical, bluish, hyperchromatic cells with scant cytoplasm. Admixed are cords and nests of enlarged, epitheloid cells with abundant, pale cytoplasm, uniform, spheroidal nuclei and vesicular chromatin. The cytoplasm is impacted with melanin pigment although focal pigment granules can be identified. Tumour cells are disseminated within a dense, fibrotic background [4, 5].
Microscopically, melanotic neuroectodermal tumour of infancy is composed of islands of miniature, spherical neoplastic cells embedded amidst a fibro-collagenous stroma. Characteristic histological feature is a biphasic tumour cell population constituted of enlarged, epitheloid, melanogenic or melanin-producing cells and miniature, primitive, neurogenic or neuroblast-like cells. Tumour cells configure sheets, nests, cords, pseudo-glandular arrangements or occasional pseudo-alveolar spaces. The articulations are composed of an admixture of enlarged, melanogenic cells and miniature, neurogenic cells although ascribed cellular populations can appear singularly [5].Generally, melanogenic cells are delineated within the periphery whereas spheroidal neurogenic cells accumulate within centroidal region, configuring cellular nests or tumour aggregates.Melanogenic cells are moderate to enlarged, appear as epitheloid, polygonal or cuboidal, imbued with abundant eosinophilic cytoplasm and melanin granules. Nuclear chromatin is smooth to vesicular with occasional, prominent nucleoli [5, 6].Neurogenic population of tumour cells is comprised of miniature, primitive cells with minimal cytoplasm and elevated nuclear to cytoplasmic ratio. Nuclei are spherical, hyperchromatic and occasionally demonstrate salt and pepper chromatin. Neurofibrillary stroma enveloping neurogenic cells can be discerned in certain tumours. Tumour cells are intermixed with well vascularized, dense, fibro-collagenous stroma. Typically, mitosis and tumour necrosis are exceptional and accompany obviously malignant biological behaviour. Periphery of tumour aggregates localized within bone is infiltrative and nests of tumour cells are situated betwixt bony trabeculae. Tumour evolution with invasion of miniature neurogenic cells can be misinterpreted as a malignant neoplasm [5, 6]. As melanotic neuroectodermal tumour of infancy is composed of miniature, spheroidal, neuroblast- like tumour cells in combination with enlarged, polygonal, melanin- containing cells, it enunciates an amalgamation of neural, melanocytic and epithelial cells. Aforesaid heterogeneous cellular subtypes are incurred due to mesodermal and ectodermal morphological manifestations engendered by neural crest within diverse phases of ontogeny. M2- polarized macrophages contribute towards pathogenesis of neoplasm [6].
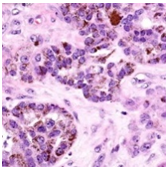
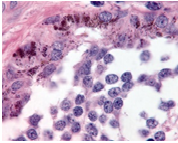

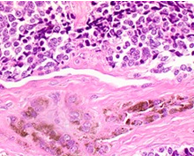

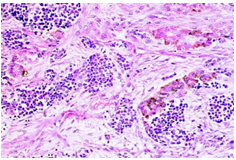
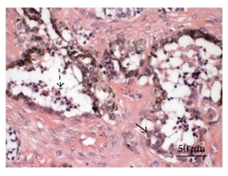
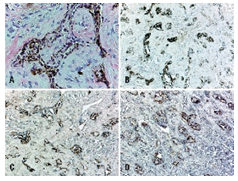
Immune Histochemical Elucidation
Miniature, round, primitive, neuroblast-like tumour cell component is immune reactive to synaptophysin, neuron specific enolase(NSE), glial fibrillary acidic protein (GFAP) and CD57. Enlarged, epitheloid, melanin-producing tumour cells are immune reactive to anti-cytokeratin monoclonal antibodies (AE1/AE3) and human melanoma black 45 (HMB45) antigen. Dendritic-like cells, immune reactive for HLA-DR, Factor XIIIa, CD68, CD163 can be observed within glia- like tissue, fibrous tissue septa and within tumour stroma. Aforesaid cells can be implicated in modulating tumour growth and/ or remodelling of intervening tumour stroma[6,7].
Immune markers such as human melanoma black 45 (HMB45) antigen, Melan A, cytokeratin along with neuroblastic markers as synaptophysin, neuron specific enolase (NSE) can assist in discerning the neoplasm. Melanin-producing epithelial cells are intensely immune reactive to HMB45 and Melan A [6, 7].Melanogenic and neurogenic cells comprising melanotic neuroectodermal tumour of infancy are immune reactive to vimentin and neuron specific enolase(NSE). Enlarged epitheloid, melanogenic cells are commonly reactive for cytokeratin and markers of melanocytic differentiation as human melanoma black 45 (HMB45) antigen and dopamine β-hydroxylase. Enlarged epitheloid cells are variably immune reactive for synaptophysin. A proportion of melanogenic cells are immune reactive to epithelial membrane antigen (EMA) [6, 7].Miniature neurogenic cells are immune reactive to synaptophysin and non-reactive for cytokeratin. Neuroblast- like cells can occasionally be immune reactive to glial fibrillary acidic protein (GFAP) although immune reactivity to neurofilament and CD99 is exceptional.Melanogenic and neurogenic cells are typically non-reactive to chromogranin and S100 protein with few instances delineating a focal expression.Foci of glial differentiation and varying stages of skeletal muscle can be enunciated in few instances with immune staining for myogenic markers such as desmin, muscle specific actin (MSA) or myogenin. Aforesaid immune reactivity can exemplify a misrepresentation as rhabdomyosarcoma[7,8].
Melanotic neuroectodermal tumour of infancy can express histological and immune phenotypic features akin to adjunctive lesions such as cellular blue nevus, malignant melanoma, neuroblastoma or rhabdomyosarcoma although diffuse reactivity with S100 protein is not exemplified.On ultrastructural examination, melanogenic cells are encompassed by basal lamina and configure desmosomes with adjacent cells whereas neurogenic cells depict dense vesicles [6, 8].
Differential Diagnosis
Melanotic neuroectodermal tumour of infancy arising in head and neck requires segregation from Ewing’s sarcoma, lymphoma, odontogenic tumours, developmental cysts, rhabdomyosarcoma, metastatic neuroblastoma, localized infection and non odontogenic lesions as fibromatosis or fibrous dysplasia [3].Melanotic neuroectodermal tumour of infancy mandates distinction from adjunctive childhood round cell tumours such as neuroblastoma, Ewing’s sarcoma, alveolar rhabdomyosarcoma or lymphoma. Also, melanogenic tumours such as malignant melanoma or clear cell sarcoma of soft tissue necessitate demarcation [3, 4].Distinctive features enunciated by melanotic neuroectodermal of infancy are cogent clinical representation, biphasic population of epitheloid, melanogenic cells admixed with primitive, neurogenic cells along with characteristic immune reactive panel.Evaluation of miniature tissue specimens can be challenging as the neoplasm can be misdiagnosed as a neuroblastoma[3,4].Olfactory neuroblastoma is frequently discerned within fifth to sixth decade. Typically, cribriform plate, nasal concha or septum is incriminated. Tumour cells are miniature, spheroidal, bluish and configure Homer-Wright rosettes, falsely imparting a biphasic cellular morphology although constituent cells are of singular subtype. Tumour cells are immune reactive to neuron specific enolase (NSE) and synaptophysin, akin to primitive neurogenic cells of melanotic neuroectodermal tumour of infancy. Also, neuroblastoma lacks a population of epitheloid melanogenic cells imbued with melanin pigment and is non-reactive to cytokeratin or HMB45[3,4].However, melanotic neuroectodermal tumour of infancy is typically devoid of rosette articulations and diffuse immune reactivity to neuroendocrine markers.An estimated 2% to 10% of Ewing’s sarcoma appear within head and neck. Ewing’s sarcoma is comprised of nests and sheets of miniature, monomorphic, spherical cells. Cellular component can recapitulate neuroblast- like cells of melanotic neuroectodermal tumour of infancy wherein enlarged, pigmented, epitheloid melanogenic cells are absent. Tumour cells depict an intense, diffuse, cytoplasmic immune reactivity to CD99. The neoplasm typically harbours chromosomal rearrangements of EWSR1 gene. Majority of Ewing’s sarcoma depict chromosomal translocation t(11;22)(q24;q12) with EWSR1-FLI chimeric fusion. Although melanotic neuroectodermal tumour of infancy can exceptionally react to CD99, absence of molecular signatures of Ewing’s sarcoma can be beneficially adopted for demarcating the neoplasms [3, 4].Alveolar rhabdomyosarcoma is commonly discerned within extremities although head and neck region is often incriminated. The neoplasm is constituted of nests of poorly differentiated, miniature, hyperchromatic cells subdivided by fibrous tissue septa. Tumour cells of alveolar rhabdomyosarcoma are focally immune reactive to desmin and display a diffuse, intense reactivity to myogenin. Characteristic chromosomal translocation t(12;13) or t(1;13) along with fusion of PAX-FOX01 gene is denominated. As melanotic neuroectodermal tumour of infancy can demonstrate focal muscular differentiation, molecular assay and fluorescent in situ hybridization (FISH) evaluation is advantageous for appropriate distinction [3, 4].Rhabdomyosarcoma is a neoplasm occurring in older individuals whereas embryonal rhabdomyoblastoma is a frequent subtype incriminating younger children. The neoplasm commonly implicates sinonasal tract and is composed of miniature, primitive, spherical, bluish cells intermingled within scattered rhabdomyoblasts. The neoplasm is immune reactive to myogenic markers such as myogenin or myoD1. Epitheloid, pigmented, melanogenic cellular component is usually absent [3, 4].Ewing’s sarcoma and alveolar rhabdomyosarcoma delineate extensive tumour necrosis, a feature typically absent in melanotic neuroectodermal tumour of infancy.Benign or malignant haemato-lymphoid cells can morphologically simulate neurogenic cells although an expression of haemato-lymphocytic markers is observed along with non-reactivity to epithelial or melanotic immune markers(3,4).Immune profile of melanotic neuroectodermal tumour of infancy is diverse from lymphoma. Also, the neoplasm is devoid of reoccurring molecular modifications. Thus, cogent molecular and cytogenetic analysis is beneficial in differentiating the neoplasms with characteristic chromosomal translocations accompanying haemato-lymphoid neoplasia[4].
Approximately 16% of non –Hodgkin’s lymphoma (NHLs) are discerned within jaw bones. An estimated two-thirds instances are diffuse large B cell lymphoma (DLBCL). Older age individuals are commonly implicated. Lymphoblastic lymphoma or Burkitt’s lymphoma predominantly demonstrate monotonous, miniature to medium- sized spherical cells in the absence of biphasic cell population. Flow cytometry, immune histochemical assay for terminal deoxynucleotidyl transferase (TdT) and molecular studies can assist tumour differentiation and classification.Malignant melanoma appearing in childhood delineates a distinctly malignant cellular morphology, is immune reactive to S100 protein or adjunctive, immune reactive melanoma markers and is non-reactive to cytokeratin[3,4].Classically, clear cell sarcoma occurs within extremities of young adults. The neoplasm is comprised of uniform, spindle-shaped to elliptical cells with clear to pale, eosinophilic cytoplasm. Primitive, neurogenic cells are absent. Clear cell sarcoma characteristically demonstrates reoccurring chromosomal translocation t(12;22)(q13;q12) with consequent emergence of EWS-ATF1 chimeric gene and an infrequent t(2;22)(q34;q12) genetic translocation[3,4].
Congenital granular cell tumour of new born (congenital epulis) emerges within a similar age group as melanotic neuroectodermal tumour of infancy. Nevertheless, characteristic histology aids distinction betwixt the entities[3].Odontogenic tumour of childhood can clinically simulate melanotic neuroectodermal tumour of infancy on account of identical tumour localization and representation, although the neoplasm is exceptional prior to 6 years[4].Desmoplastic small round cell tumour is an infrequent, aggressive neoplasm arising within abdomen of adolescents and young adults. A male preponderance is observed. Neoplasm is composed of nests of spheroidal, blue cells with variable cytoplasm, hyperchromatic nuclei and a circumscription of desmoplastic stroma. Epitheloid, melanogenic cellular component is usually absent. Chromosomal rearrangements of EWSR1 gene, akin to Ewing’s sarcoma, are discerned [3, 4].
Investigative Assay
Evaluation with computerized tomography (CT) is optimal for tumour discernment besides procuring information regarding appropriate surgical manoeuver. Intraosseous lesions commonly display a well circumscribed, hypodense tumefaction. Aggressive, advanced tumours are accompanied by extensive bone destruction. A well-defined, unilocular, osteolytic lesion is cogitated which expands and decimates incriminated cortical bone (7, 8). Imaging studies represent the tumour as a well demarcated, radiolucent, bony, lytic lesion with accompanying features of localized demolition of soft tissue, akin to a malignant neoplasm.Computerized tomography (CT) demonstrates a hyper-dense nodule with enhanced bone remodelling and expansion(7).Magnetic resonance imaging (MRI) reveals a circumscribed, expansive neoplasm appearing hypo-intense upon T1 and T2 weighted imaging.Generally, computerized tomography is adopted to define the neoplasm and evaluate appropriate surgical approach although magnetic resonance imaging is superior in illustrating the soft tissue component. Due to diverse imaging features described with melanotic neuroectodermal tumour of infancy, accrual of cogent tissue specimens is necessary for appropriate diagnosis (8).
Prognostic Outcomes Although benign, biologic behaviour of melanotic neuroectodermal tumour of infancy is inadequately inferenced. Rapid tumour progression and significant, localized tumour infiltration or relapse can be alarming. Localized tumour reoccurrence appears in an estimated 6.5% subjects although the proportion varies from 10% to >35% individuals. Tumour reoccurrence appears on account of multi-centric tumour growth or inadequate surgical extermination of the neoplasm with multiple surgical interventions. Fatality due to tumour reappearance is observed, especially with incrimination of central nervous system or vital organs [8, 9].
Malignant melanotic neuroectodermal tumour of infancy accounts for an estimated 6.5% instances and segregation betwixt benign and malignant neoplasm can be challenging as pertinent diagnostic criterion or immune markers are undefined. Cogent histological manifestations along with a mitotic index ≥ of 2 mitosis per 10 high power fields or Ki67 proliferation index exceeding >25% and immune reactive CD99 are associated with tumour aggression. Preponderant neuroblast -like cells with an insignificant component of enlarged, melanogenic cells also display aggressive behaviour and enhanced localized tumour reoccurrence. Preliminary age of disease discernment appears to be a prognostic indicator of tumour relapse. Tumours detected within 2 months of infant life can probably reappear within 6 months and are associated with reduced disease- free survival. Tumours detected within 4.5 months of infant life delineate an intermediate possibility of tumour reoccurrence. Melanotic neuroectodermal tumour of infancy is associated with tumour metastasis in an estimated 3% instances. Neoplastic dissemination occurs within lymph nodes and central nervous system [7, 9].
Therapeutic Options
Optimal therapy for melanotic neuroectodermal tumour of infancy is comprehensive surgical excision with removal of a broad, 5 millimetre perimeter of uninvolved tissue along with eradication of incriminated teeth [8, 9].Comprehensive, localized surgical eradication of the neoplasm with tumour- free surgical perimeter is preferred. Adjuvant therapy can be adopted for treating residual neoplasm or tumour reoccurrence.Singular chemotherapy or radiation therapy or combination of chemotherapy with surgical extermination of the lesion can be adopted. The lesion can be subjected to enucleation and curettage. Adopted therapeutic modality is contemplated as curative where the neoplasm can be severed from abutting bone. Thereby, inferior aesthetic outcomes and altered facial development can be circumvented [8, 9].Tumour reoccurrence is discerned within 6 months following treatment and generally appears in subjects below < 4.5 months. Due to rapid tumour enhancement observed with melanotic neuroectodermal tumour of infancy with consequent, possible major anatomic deformities within circumscribing tissues, antecedent diagnosis is anticipated in order to obtain superior functional and therapeutic outcomes with minimalistic mutilating surgeries[8,9].Chemotherapy and/ or radiation therapy is employed in around 9.6% instances, especially prior to surgical eradication, in order to decimate tumour magnitude and enable comprehensive tumour resection.Neoadjuvant therapy is applicable for inoperable tumours, incrimination of central nervous system and vital organs or where surgical perimeter is undefined and tumour-incriminated. Cogent chemotherapy can initiate maturation of primitive, neurogenic tumour cells, depletion of neuroblast-like cellular component and predominance of epitheloid, melanogenic component [8, 9].
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
