
Review Article | DOI: https://doi.org/10.31579/2690-1919/398
1 The Third School of Clinical Medicine, Southern Medical University, Guangzhou,510515, China,
2 Medical Department, Qingbu Community Health Service Center, Huadu District, Guangzhou, Guangzhou ,510800, China,
3 Department of Nephrology, Huadu District People’s Hospital of Guangzhou, Guangzhou,510800, China,
4 Department of Central laboratory, Huadu District People’s Hospital Guangzhou, Guangzhou,510800, China.
*Corresponding Author: Yanyan Su, Department of Nephrology, Huadu District People’s Hospital of Guangzhou, 510800,No. 48 Xinhua Road, Guangzhou, China
Citation: He Qin, Peng Rongdong, Li Zukai, Feng Junxia, Zhou Xiaoying, Su Yanyan , (2024), Targeting on the podocyte immune response in diabetic nephropathy: from pathogenesis to therapy, J Clinical Research and Reports, 16(5); DOI:10.31579/2690-1919/398
Copyright: © 2024, Su Yanyan. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 27 August 2024 | Accepted: 18 September 2024 | Published: 30 September 2024
Keywords: podocyte; immune response; diabetic nephropathy
The dysfunction of the glomerular filtration barrier (GFB) is a main feature of diabetic nephropathy (DN). Podocyte, terminally differentiated epithelial cell, is located on the outer of the glomerular basement membrane and play a pivotal role in maintaining the integrity of GFB. Damage in podocyte leads to proteinuria in the early stages of DN and eventually develops into chronic kidney disease. Podocyte expresses certain immune characteristics and this may predispose them to immune injury in diseases. Immune-mediated podocyte injury is a vital pathogenic mechanism in DN. Numerous evidence indicate that podocytes serve as a target of immune response in both innate and adaptive immunity, which mediate the podocyte injury in DN. In this mini-review, we focus on recent developments involving in the immune mechanisms of podocyte injury in DN and provide recent advances in the therapeutic strategy of podocyte protection. This review offers valuable opinions for understanding immune-related mechanisms in podocyte injury and identification of potential therapeutic targets, which have significant implications for future research and clinical treatment.
Diabetic nephropathy (DN) is one of microvascular complications in diabetes mellitus, accounting for the major cause of end-stage renal disease (ESRD) worldwide [1]. Increasing evidence indicate that glucose-lowering agents have limited efficacy in preventing the progression of DN in its later stages. The clinical development of DN is classified according to the level of urine albumin excretion and the decline in glomerular filtration rate (GFR). In the development of DN, urine albumin to creatinine ratio (UACR) between 30 to 299 mg/g and UACR ≥300 mg/g are separately described as incipient nephropathy and overt nephropathy, and GFR <30>
It has been well known that complex mechanisms are involved in the pathogenesis of DN onset and development. Podocyte injury is one of the main characteristic features in DN, occurring as a consequence of their loss and dysfunction, which is a driver of albuminuria [5-6]. Podocytes account for major component of the GFB and are highly specialized epithelial cells with limited capacity for renewal, which means podocyte loss can hardly be fully compensated by regeneration nearby healthy podocytes [7]. Thus, understanding the mechanism of podocyte injury in DN can provide insights to therapeutic strategies in the future. Up to date, emerging studies show that multiple mechanisms are involved in podocyte injury, including abnormal apoptosis, pyroptosis, necroptosis, autophagy, inflammation and immune-related cell death, as well as several molecular signal pathways [8-10]. Moreover, podocyte exhibits some features of immune cell, such as expressing certain immune characteristics (MHC class, B7-1, FcRn) and acting as antigen-presenting cell [11-12], and the podocyte's properties in immunity may predispose it to immune injury in kidney diseases. In this review, we aim to determine the immune-related mechanisms of podocyte injury in DN and further discuss the agents recommended by guidelines on how best to protect the life of podocytes in our kidneys.
1 Inflammation in DN
Numerous studies illustrate that both systemic and local renal inflammation are associated with DN. In different stages of DN, elevated levels of inflammatory cytokines and immune mediators can be detected in serum and peripheral blood cells, such as interleukin-6 (IL-6), tumor necrosis factor-a (TNF-a), monocyte chemoattractant protein-1 (MCP-1), intracellular adhesion molecule-1 (ICAM-1) and myeloid differentiation factor 88 (MyD88) [13,14]. In patients with type 2 diabetes, chronic inflammation in adipose tissue is considered a vital contributor to insulin resistance [15]. The serum C-reactive protein (CRP), a marker of systemic inflammation, increases in early diabetic kidney injury and is associated with the development of microalbuminuria in diabetic patient [16]. Urine level of MCP-1 is upregulated in diabetic patient with albuminuria and relates to the decline of renal function [17]. Furthermore, the urine level of MCP-1 is considered a predictor of the progression of DN [18]. These evidence demonstrate the vital role of inflammation in the development of patients with DN.
Histological analysis of renal biopsy is considered a “golden standard” strategy to diagnose the features of DN. However, in real clinical activity, renal biopsies are rarely performed in diabetic patients. Those who have substantial increase in albuminuria or significant decline in renal function, are suitable patients to perform renal biopsies to determine whether there are additional diseases [19,20]. Due to above limitations, animal model studies have unique advantages in understanding pathological characteristics of kidney injury [21]. Animal models can partially exhibit pathological characteristics of kidney injury that are similar to patients, and these strategies help researchers to well understand the process of pathology in DN [22]. Plentiful of animal studies have illustrated inflammatory cell infiltration in the kidney is associated with the progression of DN. Proinflammatory cytokines and leucocyte adhesion molecules are elevated in diabetic kidneys, while inhibition of inflammatory response can alleviate the development of renal inflammation and improve renal function [ 23-26]. This evidence supports inflammations in the kidney, an early feature of DN, which is required for disease progression.
2 immune responses associated with podocyte injury
Podocyte injury is one of the major characteristics in DN and plays an essential role in the progression of albuminuria [1]. Emerging evidence illustrate podocyte displays the properties of immune cell and this may predispose podocyte to damage by immune response [12] Studies also demonstrate that innate and adaptive immune responses extensively exert effects in the onset and progression of podocyte injury, which supports the inseparably intimate relation between podocyte injury and immune response in DN (as shown in Figure1).
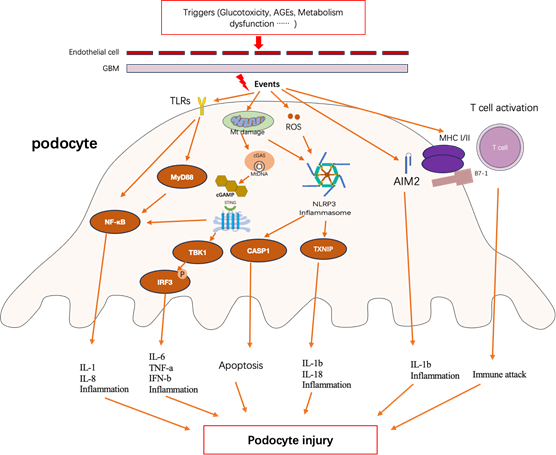
Figure 1
Innate and adaptive immune response in podocyte lead to podocyte injury in diabetic nephropathy. Triggers that induce the immune response in podocyte include various factors, including glucotoxicity, advanced glycation end-products (AGEs) and metabolism dysfunction such as lipidic toxicity [ 27-28]. Events are the form of actions when podocyte response to extracellular stress, include a range of molecular mechanism [29]. Podocyte injury contains podocyte effacement, apoptosis, cell structure alteration and cell dysfunction and so on [30]. GBM, glomerular basement membrane; TLRs, toll-like receptors; MyD88, myeloid differentiation factor 88; NF-kB, nuclear factor kappa B; Mt DNA, mitochondrial DNA; cGAS, cyclic GMP-AMP synthase; cGAMP, 2’3-cyclic GMP-AMP; STING, stimulator of interferon gene; TBK1, TNK-binding kinase1; IRF3, interferon regulatory factor 3; ROS, reactive oxygen species; NLRP3, NLR family pyrin domain containing 3; CASP1, caspase 1;TXNIP, thioredoxin (TRX)-interacting protein;AIM2, absent in melanoma 2; MHC, major histocompatibility complex; IL-1b, interleukin 1b; IL-1, interleukin 1; IL6, interleukin 6; IL-18, interleukin 18; IL8, interleukin 8;TNF-a, tumor necrosis factor alpha; IFN-b, interferon b;
2.1 Innate immune responses associated with podocyte injury
The innate immune system serves as the front defense of the host to eliminate invading pathogens [31]. Difference from the adaptive immune system, the innate immune system is capable of quickly detecting and removing the invaders without clonal expansion of antigen-specific lymphocytes. The innate immune system depends on pattern recognition receptors (PRRs), which can recognize pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs). There are various types of PRRs in mammalian animals, including toll-like receptors (TLRs), retinoic acid-inducible Gene-I (RIG-I)-like receptors (RLRs), Nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs), absent in melanoma 2 (AIM2)-like receptors (ALRs), and C-type lectin receptors (CLRs), as well as cyclic GMP-AMP synthase (cGas) and other intracellular DNA sensors [32-34]. According to subcellular localization, they can be divided into membrane-bound PRRs (TLRs and CLRs) or cytoplasmic PRRs (NLRs, RLRs, ALRs, and cGas) [35]. PRRs are mainly expressed in immune cells, such as macrophages and dendritic cells, as well as nonimmune cells, such as podocytes. Podocyte has been found to express various PRRs which play a vital role in response to innate immunity in DN [11].
2.1.1 Involvement with TLRs
The best-characterized PRRs are TLRs, which have been extensively explored. To date, studies show that ten kinds of TLRs (TLR1-10) in human species and twelve kinds of TLRs (TLR1-9, TLR11-13) in mouse species have been identified [36]. TLR located on the cell surface have the N-terminal and Toll/interleukin-1 receptor (TIR) domain, the former consists of the Leucine-rich repeat (LRR) with substrate binding ability towards the extracellular, and the latter is responsible for transferring signals into the cell [37]. Various types of cells can express TLRs, including immune cells, endothelia and epithelial cells. Podocyte has been found to express TLRs and the activation of TLRs in podocyte is the response to cell stress and injury, which can induce secretion of cytokines and chemokines to prompt the innate immune response [38-39]. For instance, in human immortalized podocytes, induction of podocyte injury by puromycin aminonucleoside is associated with the upregulation of TLR9 expression and activation of nuclear factor kappa-B (NF-kB) [40]. TLR2 and TLR4 signaling are involved in the pathogenesis of several kidney diseases, such as acute kidney disease [41], kidney transplantation [42] and diabetic nephropathy [43]. These evidence demonstrate a double role of TLRs in podocytes, not only as major players in response to foreign pathogens but also as mediators of podocyte injury. Additionally, among TLRs, preclinical and clinical studies support the causative role of TLR2 and TLR4 in DN [39], thereby in this section we mainly focus on the role of TLR2 and TLR4 in podocyte injury.
It is reported that high glucose induced the overexpression of TLR2 and TLR4 in kidney tissues, the process of which were associated with the increased levels of several chemokines and immune mediators, such as NF-kB, interleukin-8 (IL-8), MCP-1, ICAM-1, and vascular cell adhesion molecule-1 (VCAM-1) [44]. High glucose directly promoted TLR4 activation in podocytes in vitro, resulting in MyD88/NF-kB activation and inflammatory response. Consistent with this, elevated expression of TLR4 and its downstream cytokines and chemokines were detected in diabetic nephropathy in WT mice with STZ-induced diabetes [45]. High glucose also directly promoted TLR2 activation in podocytes and tubular epithelial cells in vitro, resulting in NF-kB activation and inflammation [46]. Interestingly, treatment with TLR2 agonists further exacerbated podocyte injury, inflammatory infiltration and serum creatinine levels in STZ-induced diabetic mice [47]. On the other hand, blocking the Toll-like receptor might have potential protection for diabetic podocyte damage. In cultured human podocytes with sera from T1D patients, high LPS activity induced cell apoptosis via the downregulation of the Akt cell survival pathway, however, these effects were prevented by inhibiting the TLR signaling pathway with immunomodulatory agent GIT27 [48]. Human umbilical cord mesenchymal stem cells decreased the inflammation of podocytes under high glucose via inhibition of TLR2 and TLR4 signaling pathways [49], suggesting their vital role in mediating inflammation in diabetic nephropathy. Compared to diabetic WT mice, TLR4-/- mice were protected against the development of diabetic nephropathy, exhibiting less podocyte injury, albuminuria and inflammation [45]. Similar to TLR4-/- mice, deficient TLR2 mice also displayed protection against podocyte injury in diabetic nephropathy [46]. Collectively, both genetic deletion and pharmacological inhibition of TLR2 and TLR4 in diabetic mice attenuate podocyte injury and kidney dysfunction, suggesting that they might be potential targets for podocyte protection in DN.
2.1.2 Involvement with NLRs
It has been well known that nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) are intracellular receptors, mainly located in the cytoplasm, which have C-terminal leucine-rich nucleotide-binding (NACHT) and N-terminal effector domain. NLRs can be further subdivided into four different subfamilies, including NLRAs, NLRBs, NLRCs, and NLRPs, according to their N-terminal domains. [ 50] The NLRs exert multiple effects and play pivotal roles in innate immunity, such as presenting antigens, pathogen sensing and mediation in inflammation [51]. Although the links between NLRs and renal disease still need to be further elucidated, increasing pieces of evidence demonstrate that several NLRs, including NLR family pyrin domain containing 3 (NLRP3), NOD1, and NOD2 play crucial roles in the pathophysiological process of DN [52].
NLRP3. The proteins of the NLR family are mainly localized in the cell and are divided into NLRC proteins with CARD domains and NLRP proteins with pyrin domains, according to their N-terminal domain. NLRP3 is an important member of the NLRs family, which has been studied extensively [53]. The NLRP3 inflammasome and its downstream signaling molecules, such as caspase-1, interleukin -1b (IL-1b), and interleukin-18 (IL-18), play a pivotal role in cell death and are associated with podocyte injury in DN [54]. The process of NLRP3 inflammasome activation is regulated not only by infection and inflammation but also by nutrition and metabolism [55]. It is generally thought that the activation of NLRP3 inflammasome involves two steps, including priming and protein complex assembly, the former step is triggered by PRR signaling or cytokines which leads to the transcriptional activation of NLRP3 inflammasome components, and the later step is induced by various PAMPs and DAMPs [56-57]. A few studies support that the overactivation of NLRP3 inflammasome often leads to various diseases and cell injury [58-61]. It has also been shown that inhibition of NLRP3 inflammasome leads to the suppression of multiple proinflammatory signaling pathways and improves cell survival and tissue function, supporting that NLRP3 inflammasome might be the potential target for the treatment of podocyte damage in diabetes. [62-63]
Experimentally, study from Gao et al. showed that podocytes were capable of expressing NLRP3 inflammasome, and exposure to high glucose could activate NLRP3 inflammasome and induce IL-1 production by interacting with thioredoxin (TRX)-interacting protein (TXNIP), which eventually resulted in podocyte injury [64]. Hyperglycemia induced the expression of TXNIP and subsequently caused the activation of gp91, which was a subunit of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. In vivo studies, genetic deletion of TXNIP abrogated NLRP3 inflammasome activation through inhibition of NADPH oxidase and alleviated podocyte injury in STZ-induced diabetic mice [65]. In the presence of high glucose or AGEs, mitochondrial-derived reactive oxygen species (ROS) stimulated the NLRP3 inflammasome in podocytes from diabetic patients, while mice with abolishment of NLRP3 expression in non-myeloid derived cells have protection against diabetic nephropathy [66]. Apoptosis-associated speck-like protein (ASC) is a component of NLRP3 inflammasome. Inhibition of ASC by shRNA transfection in high-fat diet mice attenuated proteinuria, foot process effacement and loss of slit diaphragm molecules in podocytes [55]. In vitro cultured podocytes, pharmacological or siRNA inhibition of NLRP3 markedly decreased high glucose-induced apoptosis, cytoskeleton change and lipid accumulation [67], as well as restoration in podocyte autophagy and nephrin expression [68].
However, Sophie et al. argued that NLRP3 was poorly expressed or virtually absent in podocyte [69]. In this study, RNA-Seq data released from the Human Protein Atlas were explored by single-cell transcriptome analysis and the results showed the absence of relevant amounts of transcripts for NLRP3, IL-1b and other related molecules. They further identified the lack of mRNA expression of NLRP3, CASP1(caspase 1) and IL-1b in cultured human podocytes, even under the presence of NLRP3 inflammasome inducers lipopolysaccharides (LPS) and adenosine triphosphate (ATP). They also raised the limitations of their study, in which the cultured human podocyte transcriptomes were not performed in diabetic conditions and the mouse model of diabetes did not develop overt diabetic nephropathy. In brief, whether podocyte express NLRP3 inflammasome seems contradictive in the published studies, suggesting that further studies are needed to identify the role of NLRP3 in podocyte injury of diabetic kidney disease.
NOD1 and NOD2. The expression of NOD2 can be identified in various types of cells, including tubular epithelial cells, glomerular endothelial cells and podocytes, and plays a crucial role in the pathogenesis of DN [70]. Clinical and animal studies demonstrate that the upregulation of NOD2 expression can be detected in the kidney biospy samples from patients with T2DM and STZ-induced diabetic mice. Experimentally, the presence of high glucose, advanced glycation end-products (AGEs), TNF-a and transforming growth factor-b (TGF-b) significantly increased and activated the NOD2 expression in vitro cultured podocytes, and the activation of NOD2 expression induced the secretion of proinflammatory and profibrotic mediators, resulting in podocyte injury [71]. RNA-binding protein human antigen (HuR) is a key posttranscriptional regulator of NOD2 expression, acting to enhance NOD2 mRNA stability. In the kidneys of patients with DN, the HuR expression was upregulated and closely related to the progression of proteinuria [72]. It had also been found that NOD2 affected glucose handling and nephrin expression in podocytes, as well as TGF-b signaling in tubular cells and immune cells [71-73]. On the other hand, genetic NOD2 knockout in diabetic mice or inhibition of NOD2 expression in cultured podocytes displayed protection against the hyperglycemia-induced podocyte damage, leading to the alleviation of loss in nephrin and disruption of actin filament structure [71]. Moreover, the activation of NOD1 expression is related to metabolism inflammation and insulin resistance induced by obesity. Study found that the NOD1-RICK-NF-kB inflammatory signaling pathway contributed to the pathogenesis and progression of DN. NOD1 deficiency had protection against high-fat diet-induced insulin resistance [74]. However, whether NOD1 is involved in podocyte injury in the development of DN needs to be further explored. Collectively, based on the above evidence, therapies targeting on NLRs may provide promising benefits for podocyte protection.
2.1.3 Involvement with RLRs
The retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) are RNA sensors, which are localized in the cytosol and act on important inducers of type I interferons and other antiviral immune mediators [75]. This protein family includes three members: RIG-I, melanoma differentiation-associated gene5 (MDA5) and laboratory of genetics and physiology 2 (LGP2). Generally speaking, RLRs consist of a central helicase and a carboxy-terminal domain (CTD), while RIG-I and MDA5 additionally have two N-terminal caspase recruitment domains (CARD). These domains work together in response to immunostimulatory dsRNA and exert crucial effects on viral infection.Moreover, RLRs are associated with a range of cellular processes, such as proliferation, differentiation and apoptosis [76]. It has been shown that besides immune cells, podocytes express RIG-I and MDA5, and the activation of RLR is related to podocyte damage. For instance, Yamashita et al. reported that in cultured human and mouse podocytes, the RLR activation in podocytes induced by dsRNA led to the expression of interferon I ( IFN-I), IL-6 and cytoskeleton alteration, eventually resulting in podocyte effacement [77]. Apolipoprotein L1 (APOL1) is closely associated with primary kidney and kidney-related pathologies, which is identified in a recent large Phenome-Wide Association Study [78].Animal study showed that long-term injections of recombinant APOL1 in mice increased expression of RIG-I in podocytes. Inhibition of RIG-I expression, either by using siRNA in vitro experiments or by adeno-associated virus short hairpins (AAV-sh) in vivo mouse experiments, decreased the expression of pro-inflammatory genes and alleviated podocyte injury and glomerular damage [79]. Similar results reported by Cristian et al. also showed that in human podocytes cultured with IFN-r, inhibition of MDA5 down-regulated the expression of APOL1 and the subsequent IFN-I response [80]. The above published studies support the notion that the activation of RLRs can contribute to Sirtuin 1However, whether RLRs exert effects or how RLRs act on podocyte in diabetes remains largely elucidated.
2.1.4 Involvement with ALRs
Absent in melanoma 2 (AIM2)-like receptors (ALRs) are mainly composed of N terminal containing pyrin domain (PYD) and C terminal containing hematopoietic interferon-inducible nuclear (HIN) domain. The AIM2 protein is highly conserved in the HIN-200 family and is recently identified as the only conserved protein in both humans and mice of all ALRs [81]. AIM2 is capable of identifying abnormal cytoplasmic dsDNA from pathogens and host cells. The activation of AIM2 initiates the assembly of AIM2 inflammasome, which is composed of AIM2, an apoptosis-associated speck-like protein containing a CARD and pro-caspase-1, and plays a pivotal role in protection against pathogen infection [82]. However, the activation of AIM2 also has harmful effects in some aseptic inflammatory diseases, such as chronic kidney disease and atherosclerosis [83,84]. Emerging studies found that the activation of AIM2 inflammasome is associated with the pathogenesis of insulin resistance in patients with T2DM and renal diseases. The mechanism of AIM2 in insulin resistance (IR) and renal diseases include promotion of chronic activation in inflammatory signaling pathway and conduction of the insulin signaling pathway. For example, in the mouse model of aldosterone-induced renal injury, overactivation of AIM2 aggravated endoplasmic reticulum stress (ERS) and inflammasome activation and fibrotic changes, while silencing of AIM2 could block inflammasome-mediated signaling pathway and relieve ERS and fibrotic changes, reducing proteinuria levels in vivo [71]. In patients with T2DM, the increased circulating mtDNA levels that were closely related to mitochondrial dysfunction can be detected and are associated with AIM2 inflammasome-mediated chronic inflammation. The activation of AIM2 inflammasome elevates the expression of IL-1b, promoting IR and T2DM development [85]. However, there is an argument on the harmful role of AIM2. Gong Z et al. showed that in animal experiments, compared to WT mice, AIM2-/- mice tended to obesity and decreased energy expenditure, showing impaired brown fat function and increased fasting blood glucose and insulin resistance. The mechanism might be attributed to the AIM2 inhibition on the encoded protein 202, leading to the reduction of monocyte infiltration and lipogenesis, thus improving obesity and IR [86]. To date, there are few studies on AIM2 and podocyte injury in DN, and the seemingly contradictory role of AIM2 in metabolism means more studies are needed to confirm the association of AIM2 with diabetes and podocyte injury.
2.1.5 Involvement with CLRs
C-type lectin (CLEC) is originally defined as a protein that binds carbohydrates in a Ca2+-dependent manner, containing soluble and membrane-bound proteins. The C-type lectin receptors (CLRs) and the related C-type lectin-like receptors (CTLRs) are distinguished by their characteristics, the former have C-type lectin-like domain (CTLD) and Ca2+ ion for glycan binding, the latter possess CTLD but lack any type of ion for ligand recognition [87]. It should be noted that the term CTLRs is also used for the broad definition containing all CLEC irrespective of carbohydrate recognition because many of these receptors' binding domain structures have not been fully elucidated [88]. CLRs can be further subdivided into three clusters based on their intracellular signaling motifs. Cluster 1 consists of activation spleen tyrosine kinase (Sky)-coupled CLRs with immunoreceptor tyrosine-based activating motifs (ITAM), such as Mincle, Dectin-1 and Dectin-2. Cluster 2 includes inhibitory CLRs with immunoreceptor tyrosine-based inhibition motif (ITIM) domains, such as DCIR. Cluster 3 contains a group of CLRs lacking typical signaling motifs, such as the Mannose receptor, DEC-205, and DC-SIGN. Upon ligand binding, CLRs stimulate intracellular cascades to induce the production of inflammatory cytokines and chemokines, consequently triggering immune response to pathogens. [89] Besides, they play essential roles in the physiological functions and pathological processes including immune homeostasis, immune defenses and immune surveillance [90].
Owing to the family's large repertoire, covering them all is beyond the scope of this review, and to date, it is difficult to make a definite conclusion on whether CLRs are protective or harmful to podocytes in kidney diseases. For instance, in a mouse model experiment, CLEC14A were expressed in podocytes and showed protection against podocyte injury via anti-inflammatory effect in mice with Adriamycin-induced focal segmental glomerular sclerosis (FSGS). In vitro cultured podocytes, overexpression of CLEC14A in podocytes have anti-inflammatory and anti-apoptosis effects through inhibition of high-mobility group box 1 protein (HMGB1) release, resulting in the suppression of NF-kB signaling and early growth response protein 1 (EGR1) signaling [91]. Dectin-1 is another classic CTLR but displays pro-inflammatory effects. Upregulated expression of dectin-1 was observed in heart tissues in diabetic mice and localized to macrophages. Deficiency of Dectin-1 in mice displayed protection against diabetes-induced cardiac dysfunction, tissue fibrosis and inflammation, which means Dectin-1 is essential for cell activation and induction of inflammatory cytokines in diabetic cardiomyopathy [92]. However, there are few studies concerning the effects of CLRs on podocytes in diabetic kidney disease. The differently displayed functions of CLRs may mean each member in the CLR family has its unique role in response to various diseases.
2.1.6 Involvement with cGAS/STING pathway
The cyclic GMP-AMP synthase and stimulator of interferon gene (cGAS/STING) pathway has recently been identified as a pivotal player in innate immunity. cGAS is a vital sensor of cytosolic DNA which can catalyze the second messenger 2'3-cGAMP, and consequently activates the endoplasmic reticulum membrane adapter STING (encoded by TMEM173), leading to the activation of TNK-binding kinase1(TBK1) and interferon regulatory factor 3 (IRF3), which in turn induces the expression of type I interferons [34]. Activation of STING also leads to the activation of NF-kB in a non-canonical mechanism and induces inflammatory cytokines expression [93,94]. In response to the abnormal leakage of cytosolic DNA, regardless from the invaded pathogen, damaged mitochondrial or genomic instability, the cGAS/STING pathway can be aberrantly activated and causes various diseases, including infectious diseases, autoimmune diseases, tumors, organ fibrosis, and neurodegenerative diseases [95-97].
The bulk of studies linking the activation of cGAS/STING pathway to kidney disease have focused on the podocyte. It has been reported that podocyte expresses components of cGAS/STING pathway at the early stage of kidney dysfunction and the activation of this pathway can cause podocyte injury [98]. For instance, in a mouse model of diabetic kidney disease (DKD), treatment with STING agonist triggered podocytes loss and resulted in susceptibility to albuminuria. Similar results in vitro cultured murine and human podocytes were observed [99]. Metabolism dysfunction in diabetes can also activate this pathway. Study from Zang et al. showed that lipotoxicity in a mouse model with DKD caused mitochondrial damage and mtDNA leakage into the cytoplasm, promoting the activation of the cGAS/STING pathway which induced the secretion of inflammatory cytokines, thereby resulting in podocyte injury [98]. On the other hand, pharmacologic inhibition or genetic deletion of this pathway protected against podocyte injury and the progression of kidney disease in mice with DKD, which supports the cGAS/STING pathway as a potential therapeutic target for DKD [99].
Several studies concern the role of cGAS/STING pathway in cell crosstalk between podocyte injury and other glomerular cell damage. Qi et al. presented evidence that in DKD-susceptible mice, mtDNA from diabetes-induced mitochondrial dysfunction and stress contributed to glomerular injury indirectly through glomerular endothelial cells (GECs) and was associated with podocytes depletion, resulting in proteinuria [100]. Casalena et al. showed that in supernatant transfer experiments, diabetic serum caused mitochondrial dysfunction and superoxide release in GECs and secreted factors from dysfunctional GECs induced podocytes apoptosis [101]. Similar phenomena in which crosstalk between podocytes and GECs, can also be found in other kidney diseases, such as focal segmental glomerulosclerosis [102] and APOL1 renal risk variants [103]. Mesangial cells (MCs) also display markers of mitochondrial damage induced by diabetes, which means the leakage of mtDNA and the subsequent activation of the cGAS/STING pathway may operate in some instances of DN, mediating with podocyte injury [104].
2.2 Adaptive immune response associated with podocyte injury
Besides their role in innate immunity, podocytes are capable of promoting the adaptive immune response. It has been known that podocytes possess properties of antigen-presenting cells (APC), such as expression of major histocompatibility complex (MHC) I/ II and B7-1 [12]. MHC I and MHC II respectively contribute to the activation of CD8+ T cells and CD4+ T cells, while the B7-1 is a co-stimulatory molecule for T cells. Interestingly, B7-1(also known as CD80) is part of the B cells and APC repertoire, providing the second signal to T cells to allow amplification of response to antigens. B7-1 also directly binds to CD28 on the T cell membrane to induce a positive co-stimulatory signal [105,106]. Podocyte’s immune properties play an essential role in podocyte injury in kidney diseases. For example, Li et al. showed that B7-1 induction was related to podocyte injury in DN, while inhibition of B7-1 by an agent could efficaciously protect podocytes through prevention of systemic T-cell activation [107]. A study from Shan et al. reported that in mouse model with ovalbumin plus IFN-r and IL-17 renal injection, increased levels of MHC-I, MHC-II and B7-1 were expressed in podocytes. Podocytes could uptake and process ovalbumin, then presented the ovalbumin peptide on the cell surface, and consequently recruited specific T cells and activated T cell proliferation and inflammatory cytokine secretion, which in turn resulted in podocyte injury and developed nephropathy [108]. Li et al. presented that renal CD8+ tissue-resident-memory T (CD8+TRM) cells were activated through the interleukin-15 (IL-15) signaling pathway and played a crucial role in mediating local immune response in diabetic kidney disease. Targeting on CD8+TRM cells by pharmacological inhibition of IL-15 signaling protected against podocyte injury [109]. A study performed by Chen et al. showed that in the kidney of patients with crescentic glomerulonephritis, CD8+ T cells invaded the disrupted Bowman's space and directly contacted podocytes, resulting in podocyte destruction. However, in a healthy mouse model with an intact Bowman's capsule, the infiltration of CD8+ T cells was prevented from interacting with podocytes, suggesting that the normal Bowman's space provided a protective niche for podocytes from cytotoxic CD8+ T cells and thereby protected renal function [110]. This study may partly explain the reality that the use of reno-protective agents in clinical activity always has prominent effects against proteinuria and renal dysfunction in the early stage, but not in the end-stage kidney disease. A recent study using pod I-Ppol mice as a model showed that DNA damage in podocytes caused alterations of DNA methylation in blood cells and was associated with the proliferation of CD8+ memory T cells in the kidney, leading to proteinuria and glomerulosclerosis, which suggests the presence of crosstalk between podocyte injury and cytotoxic T cell [111]. The above evidence suggest the adaptive immune response systemically and locally contributes to the immunopathological process of podocyte injury in DN.
2.3 Involvement with Sirtuin family [112-116]
Silent information regulator 2-related enzymes (Sirtuin or SIRT) are evolutionarily conserved class III histone deacetylases, which depend on nicotinamide adenine dinucleotide (NAD+) [117]. Studies have shown that there are seven members (Sirtuin1-Sirtuin7) of the sirtuin family in mammals, of which Sirtuin1 is widely expressed in podocytes [118] and plays a critical role in mediation of cell apoptosis [119], autophagy [120], energetic homeostasis [121], mitochondrial biogenesis [122] as well as immune response [123]. Recent report by Mourits et al. illustrated the closed association of Sirtuin1 and immunology. Sirtuin1 genetic polymorphisms could affect inflammatory cytokine production of human peripheral blood mononuclear cells (PBMCs) in response to various stimuli via modulation of gene transcription [124]. Study from Chen et al. showed that Sirtuin1 was involved in the regulation of cell differentiation, activation and function in innate and adaptive immune cells [125]. Those evidence suggest Sirtuin1 might be relevant to the immune related mechanism in podocyte injury. Experimentally, Q et al. presented that in diabetic OVE26 mice with established albuminuria, overexpression of Sirtuin1 in podocytes attenuated the progression of diabetic glomerulopathy. Treatment with selective Sirtuin1 agonist in vitro cultured human podocytes protected against high glucose-induced mitochondrial injury by deacetylation of PGC-1α and activation of PPARγ-targeted gene expression, and in OVE26 mice also showed a similar result, displaying a marked reduction of albuminuria and glomerular injury [126]. Another study performed by Jiang et al. showed that in aldosterone-induced podocyte injury mice model, Sirtuin1 protected renal function through suppressing the activation of NLRP3 inflammasome, showing the greater urinary albumin excretion in podocyte-specific Sirtuin1 knockout mice than that in wild-type mice. In the cultured podocytes treated with aldosterone, overexpression of Sirtuin1 inhibited NLRP3 activation and protected against podocyte injury [127]. In addition, Sirtuin1 was also found to have renoprotective effects through improvement of the organization of actin cytoskeleton [128] and insulin resistance [129] in podocytes. Collectively, Sirtuin1 might be a potential target for podocyte protection in DN, however, more studies are needed to further elucidate the role of Sirtuin1 in the immune related podocyte injury in DN.
3 Mechanisms of agents for DN: especially focus on podocyte protection
Recently, published guideline for diabetes from the American Diabetes Association (ADA) recommend the antidiabetic agents [130]. In this guideline, sodium-glucose cotransporter 2 (SGLT2) inhibitor and glucagon-like peptide 1 receptor agonist (GLP-1RA) have direct renal protection beyond the glucose-lowering effect. ACE inhibitor (ACEI) or angiotensin II receptor blocker (ARB) are suggested for diabetic patients with hypertension, except for those who are normotensive with or without high albuminuria. Because evidence from two long-term, double-blind studies demonstrate no reno-protective effect of either ACEI or ARB among patients with type 1 and type 2 diabetes who are normotensive. Finerenone is currently the only nonsteroidal MRA with proven clinical kidney and cardiovascular benefits. The following section discusses the mechanism of these agents, especially focusing on the protection of podocyte in diabetes (as shown on table 1).
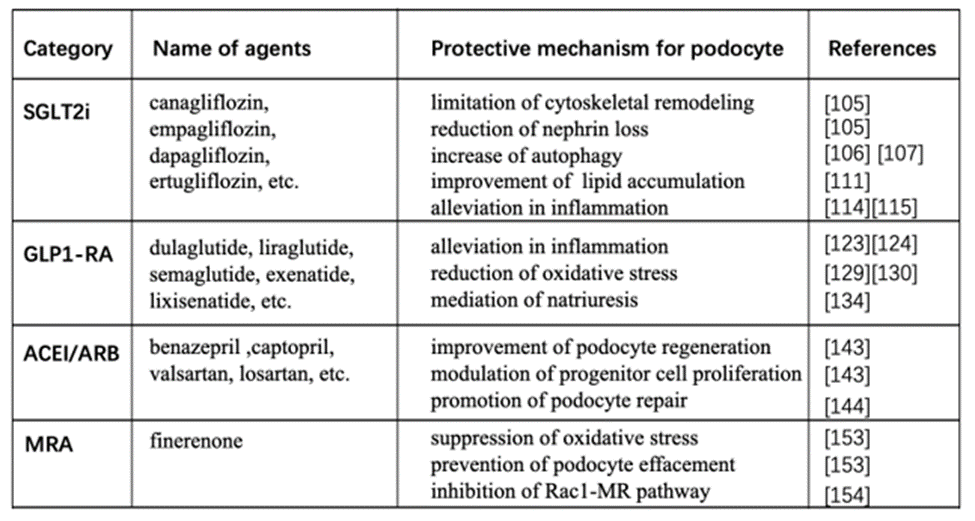
SGLT2i, sodium-glucose cotransporter 2
GLP1-RA, Glucagon-like peptide 1 receptor agonist
ACEI, Angiotensin-converting enzyme inhibitor
ARB, angiotensin II receptor blocker
MRA, Mineralocorticoid receptor antagonist
Table 1: Mechanisms of agents recommended for diabetic nephropathy by guideline
3.1 SGLT2 inhibitor
The balance between glomerular filtrate and reabsorption is one of the pivotal physiologic functions of the kidney. In euglycemia with normal GFR, the proximal tubules reabsorb almost all tubular nutrients, including glucose [131]. The bulk of glucose uptake occur in the early proximal tubule which contain high-capacity sodium-glucose cotransporter 2 (SGLT2). It has been shown that SGLT-2 accounts for all glucose reabsorption in the early proximal tubule and nearly 97% of total renal glucose reabsorption [132]. SGLT2 inhibitors are a class of antihyperglycemic drugs approved for T2DM. These drugs inhibit renal glucose reabsorption in the early proximal tubule, hence increasing urinary excretion and lowering blood glucose. Several large-scale clinical trials confirmed that SGLT2 inhibitors have pleiotropic properties beyond blood glucose-lowering effects, including beneficial renal and cardiac effects [133]. Multiple mechanisms contribute to the reno-protective effects of SGLT2 inhibitors in T2DM, such as restoration of tubule-glomerular feedback to reduce glomerular hyperfiltration, protection against inflammation, hypoxia, oxidative stress and fibrosis [134]. Direct action on podocyte is another mechanism for reno-protective function by SGLT-2 inhibitors [135].
In recent years, the expression of SGLT2 has been found in podocyte and increase in diabetic mice. For example, Cassis et al. showed that in a mouse model with proteinuria induced by bovine serum albumin (BSA), the expression of SGLT2 was upregulated in kidney after BSA injection. SGLT2 inhibition with dapagliflozin reduced the expression of SGLT2 in podocytes and protected against podocyte dysfunction through the limitation of cytoskeletal remodeling and loss of nephrin, supporting a potential mechanism that SGLT2 inhibitors directly participated in maintaining the structure and function of podocytes to prevent the progression of DN [136].
The downregulated level of autophagy in DKD has been observed and is associated with increased albuminuria and kidney dysfunction. For example,in a mouse model of type 2 diabetes performed by Anton et al, SGLT2 inhibitor empagliflozin enhanced podocyte autophagy level, showing the increased volume density of autophagosomes and autolysosomes in podocytes, thus attenuating podocyte effacement and urinary albumin excretion in db/db mice [137]. A vitro cell experiment presented by Lei et al. also illustrated a similar result. The cultured podocytes induced by advanced glycation end products (AGEs) displayed inhibition of autophagy, showing the reduced markers of autophagosome formation such as light chain 3 phospholipid conjugate (LC3II) and Beclin-1, while treatment with SGLT2 inhibitor dapagliflozin restored the expression of Beclin-1 and LC3II, promoting autophagosome turnover and autophagosome degradation, thus exerted a protective effect on podocytes [138]. These observations support that SGLT2 inhibitors exert protective effects in podocyte via the mediation of cell autophagy.The relationship between dyslipidemia and podocyte injury is well documented. Indeed, Apolipoprotein-1 (APOL1) which is considered as a high risk genetic variant in kidney disease, is highly expressed in podocyte [139]. Analysis from experimental studies and renal biopsies of patients with DN showed that sterol regulatory element-binding proteins (SREBPs), essential transcription factors associated with lipid metabolism, are increased in lipid droplets loaded podocytes [140]. In this regard, podocyte injury could be induced by lipid imbalance. SGLT2 inhibitors can improve podocyte lipid content to maintain podocyte health. Mengyuan et al. reported that in an experimental mouse model with Alport syndrome, SGLT2 inhibitor empagliflozin inhibited the utilization of glucose and pyruvate as metabolic substrate, leading to the reduction of podocyte lipotoxicity and renal cortical lipid deposition, thus improving kidney function and prolonged mice survival [141]. Dong et al. described that in mice with Western diet-induced obesity, SGLT2 inhibitor dapagliflozin decreased lipid accumulation in podocytes, which was associated with decreased SREBP-1c expression [142]. These observations support the role of SGLT2 inhibitors in the improvement of lipid metabolism in podocyte.
It is well known that low-grade inflammation is one of vital features of diabetic kidney disease. Several clinical trials illustrated that SGLT2 inhibitors have demonstrated the downregulation of circulating inflammatory markers such as IL-6, TNF-a, and IFN-r in diabetic patients [143,144]. Experimentally, in vitro cultured macrophages isolated from type 2 diabetic patients, SGLT2 inhibitor empagliflozin suppressed the secretion of IL-1b and the activation of NLRP3 inflammasome [145]. Treatment with SGLT2 inhibitor canagliflozin for 2 years in diabetic patients illustrated a decrease in levels of genes encoding TNF receptor1 and fibronectin1, which supported that SGLT2 inhibitors might alleviate molecular processes related to inflammation [146]. In line with this proposal, a study from nephrectomy rats with established CKD, SGLT2 inhibitor empagliflozin suppressed fibrosis-promoting M2 macrophage polarization and inhibition of profibrotic marker, thereby improving kidney function [147].
The above evidence support that SGLT2 inhibitors exert protective effects on podocyte through several key aspects, including maintenance of cell integrity, upregulated autophagy, improved lipid deposition and decreased inflammation. However, whether SGLT2 inhibitors have direct immune effects on mediation of podocyte injury in diabetes remains largely elucidated, thereby they are worthy of exploration in the future.
3.2 GLP-1RA
Glucagon-like peptide-1(GLP-1) is a thirty-amino acid peptide, which is released from gut enteroendocrine cells in response to the stimulation of luminal nutrients after meal ingestion. It is well known that GLP-1 exerts metabolism actions in maintaining glucose homeostasis mainly via secretion of insulin and inhibition of glucagon release as well as weight loss [148]. Glucagon-like peptide 1 receptor agonists (GLP-1RAs) are commonly used in the treatment of patients with type 2 diabetes. Numerous evidence suggest that GLP-1RAs have multiple benefits to protect the function and structure of the kidney in diabetes in addition to their glucose-lowering effects [149]. Renal outcomes observed in recent large trials showed that GLP-1RAs reduced urinal albumin excretion and weight loss, which are associated with improved survival [150-152]. Studies in models of T2DM and/or obesity illustrated that GLP-1RAs exert anti-inflammatory, anti-oxidative stress and mediate natriuresis to prevent the progression of DN and podocyte injury.
Inflammatory disorder drives the development of diabetes and its complications. A study conducted in 4213 people showed that high-sensitivity C-reactive protein (hs-CRP), a marker of systemic inflammation, is elevated in patients with T2DM [153], while an analysis from systematic review illustrated that treatment with GLP-1RAs is associated with a significant reduction of CRP, as well as other biomarkers of inflammation [154]. Anti-inflammatory activities can be found in the treatment with GLP-1RAs, including direct effects on leukocyte activation, macrophage polarization, NLRP3, and NF-kB dependent signaling pathway [155]. It has been illustrated that podocyte expresses GLP-1 receptor and GLP-1RA can act on podocyte to exert anti-inflammatory effects and prevent podocyte injury in diabetes [156]. For example, Li et al. presented that in the cultured podocytes treated with high glucose, GLP-1RA liraglutide increased podocyte survival by inhibiting the NLRP3 inflammasome pathway and reducing the secretion of IL-1b and IL-18 [157]. Moreover, studies also showed that GLP-1RA liraglutide inhibited NF-kB activation mediated by TNF-a in podocytes of obese mice and could induce distinct transcriptional changes in podocytes through inhibition of the receptor for AGEs-induced inflammation [158,159].
Oxidative stress plays a pivotal role in the mechanism of podocyte injury and DN. Numerous pieces of evidence show that GLP-1RAs have antioxidative activities independent of lowering glucose. For instance, a pilot study performed in patients with type 2 diabetes displayed that treatment with GLP-1RAs was related to reduced circulating markers of oxidative stress, such as lipid hydroperoxides, nitro-tyrosine and malondialdehyde, as well as downregulated NADPH oxidase and endogenous superoxide generation [160]. It is reported that reactive oxygen species (ROS) production through NADPH oxidases damage podocyte in diabetes, which contribute to the alterations of nephrin and podocalyxin on the podocyte surface and resulte in glomerular filtration dysfunction. ROS production in the kidney is due to the action of NOXs, including NOX1, NOX2, and NOX4. Experimentally, in vitro high glucose cultured podocytes, inhibition of the mammalian target of rapamycin (mTOR) could decrease the expression of NOX1 and NOX4 and podocyte apoptosis [161]. Moreover, targeting NOX4-derived H2O2 could alleviate podocyte injury in DN through elevation of podocyte calcium, which was associated with transient receptor potential channel 6 (TRPC6) dependent calcium influx [162]. GLP-1RA liraglutide selectively inhibited TRPC6 expression and subsequently reduced ROS production and protected podocyte structure and function in the DKD rat model [163]. In clinical studies, treatment with GLP-1RA liraglutide for 12 weeks in patients with DN increased the urine level of glutathione peroxidase, an important intrinsic antioxidant enzyme that eliminates oxidative stress [164].
Mediation in natriuresis shows another mechanism by which GLP-1RAs have renal-protective effects in diabetes. Natriuresis is the increased urinary excretion of sodium, which can result in a reduction in blood volume and a decreased workload on the heart. In mechanism, GLP-1RAs phosphorylate and inactivate the Na+/H+ exchanger 3 in the proximal tubule, consequently decrease Na+ reabsorption and promote natriuresis [165]. GLP-1RAs can also promote the secretion of atrial natriuretic peptide (ANP) in cardiac cells to increase urinary excretion of sodium and water [166]. In summary, GLP-1RA is capable of exerting protective effects on podocyte through various factors and it is necessary to further explore its immunity-related mechanism on podocyte.
3.3 Acei/Arb
The functions of the renin-angiotensin-system (RAS) have been well documented in both blood pressure regulation and renal disease development. Angiotensin II (Ang II) is the major bioactive product produced by RAS with the aid of a sequence of enzymatic processes. It exerts various physiologic effects to maintain cell and organ function, including renal cell growth, mitogenesis, apoptosis, migration and differentiation. Moreover, Ang II also induces the activation of multiple intracellular signaling pathways that are involved in renal damage [167,168].
Angiotensin-converting enzyme inhibitor (ACEI) and angiotensin II receptor blocker (ARB) have been used to control hypertension in clinical treatment. Dates from several large clinical trials have shown that in addition to lowering blood pressure and cardiovascular protective effects, ACEI and ARB have impressively reno-protective benefits and have been recommended for the treatment in patients with DKD by several guidelines [169,170]. The mechanisms of reno-protection include anti-inflammatory and anti-fibrotic effects, downregulation of sympathetic activity and reduction of aldosterone secretion.
It has been found that podocyte expresses functional components of the RAS, such as angiotensinogen, renin, ACE type1 and the AT1 and AT2 angiotensin receptor subtypes [171]. It is reported that increased Ang II is a vital risk factor for the progression of renal disease and is associated with the reduction of nephrin and podocin expression in podocyte. The loss of nephrin and podocin is causative for podocyte damage, resulting in marked albuminuria and increased podocyte apoptosis [168,172].ACE inhibition and the angiotensin blockade in the form of ACEI and ARB have been proven to be useful in preventing the progression of DN and podocyte dysfunction. For instance, Zhu et al. showed that in adult rat model with glomerularsclerosis, treatment with long-term (24 weeks) ARB significantly improved the podocyte density in glomeruli through the improvement of podocyte regeneration from parietal epithelial cells transition, as well as leading to reduction of proteinuria and prolonged rat survival [173]. Another study performed by Kelly et al. displayed a similar result in an ob/ob mouse model with diabetic nephropathy, treatment with ARB increased glomerular podocyte number and lowered proteinuria compared with diabetic ob/ob controls [174]. Research also demonstrated that ACEI therapy could sustain glomerular repair and improve kidney function via the promotion of podocyte repair, limitation of crescent generation, and modulation of progenitor cell proliferation [175]. Taken together, these findings strengthen the concept that ACEI and ARB have beneficial effects on podocyte protection and kidney function, but the mechanisms involved in DN require a multi-pronged strategy.
3.4 Mineralocorticoid receptor antagonists (MRA)
The aldosterone, the physiological main ligand of the mineralocorticoid receptor (MR), is the final component of the renin-angiotensin-aldosterone system (RAAS). Aldosterone and MR systems have been considered pivotal players in the regulation of fluid volume, electrolytes, hemodynamic homeostasis and blood pressure. MR is a subfamily of nuclear receptors that belongs to the steroid hormone receptors family, acting as intracellular receptors and nuclear transcription factors [176]. Accumulating studies implicate the abnormal reaction in aldosterone and MR system is a pathogenic factor in kidney injury independently of its physiologic functions, and the blockade of MR can prevent the detrimental effects on cell injury [177]. MR antagonists (MRAs) elicit benefits for CKD through the inhibition of aldosterone and the MR system [178]. The MR antagonists (MRAs) include steroidal MRAs and non-steroidal MRAs. In the past years, spironolactone and eplerenone were the earlier steroidal MRAs that are still widely used to date. These steroidal MRAs have been proven their benefits and utilities in heart failure and proteinuria in patients treated with an RAAS blockade by landmark trials. However, these agents remain underutilized in patients with CKD, because of the concern for hyperkalemia and worsening GFR [179].
Finerenone is a non-steroidal MR antagonist (MRA) with great cardiac and renal benefits and less effect on inducing hyperkalemia which is proven by landmark trials [180]. Finerenone is developed using a special chemical structure and optimized to create a bulky MRA without any activity at the L-type calcium channel, which may exert its effects more efficiently than a steroidal MRA [181]. The recently published guidelines from the American Diabetes Association (ADA) and the American Association of Clinical Endocrinology (AACE) recommended the addition of finerenone to standard treatment in patients with DKD [182,183]. In mechanism, the biological actions of aldosterone are mainly through MR. MR expression is present in podocyte and aldosterone appears to have direct deleterious effects on podocyte. Animal research found that rats continuously infused with aldosterone increased ROS production through NADPH oxidase and induced podocyte foot process effacement. Treatment with MRA suppressed the oxidative stress and prevented podocyte effacement [184]. Moreover, in the type2 diabetic mouse model with DN progression, the activation in RAS-related C3 botulinus toxin substrate 1 (Rac1) and MR pathway contributed to podocyte injury, while treatment with finerenone inhibited Rac1-MR pathway and protected against podocyte injury [185]. These evidence support finerenone as a vital therapeutic method in the inhibition of DN development and podocyte injury.
Conclusion
Podocyte plays a crucial role in renal physiology and pathology. For years, the exploration of mechanisms in DN has illustrated the potential for the discovery of innovative therapeutic targets. Due to the podocyte's location in the last barrier of the glomerular capillary, it has been considered as a valuable target to prevent proteinuria in DN, thereby studies focusing on podocyte protection have attracted extensive attention worldwide. Therapy aimed at preventing or limiting podocyte injury have ever been considered as a promising strategy with major potential clinical and economic benefits. Recently, the therapeutic options for podocyte protection mainly include immunosuppressive drugs and novel biological agents. However, immunosuppressive drugs, such as glucocorticoids and calcineurin inhibitors, primarily target immune-mediated inflammation in the kidney, their use may lead to serious side effects, especially after prolonged use, such as infection, hypertension and osteoporosis [186]. Novel biological agents known for its high specificity, minimal side effects, and durable benefits is developed for the treatment of podocyte disease. They also have side effects that can't be ignored including infusion response, production of anti-drug antibodies, immunogenicity and hypogammaglobulinemia. Furthermore, the optimization of parameters for immunotherapy need to improve its clinical applicability and effectiveness [187]. To date, it is still hard to exactly outline the mechanisms of podocyte injury in DN, and agents specifically targeting on podocyte protection have not been fully developed for patients with DN in clinical treatment.
As described earlier in this review, numerous preclinical studies support the promising potential of podocyte protection, especially focusing on immune-related therapies. But moving from proof of concept to clinical evaluation still has a long way to go. The molecular mechanisms of podocyte injury in DN continue to be explored, because except for immune-related injury, glucotoxicity, lipotoxicity and renal hemodynamic change in diabetes also play vital roles in podocyte injury, and the complicated crosstalk between these factors in DN may exist. These obstacles make researchers difficult to develop agents specially targeting on podocyte protection. Hopefully, further studies will provide more insights into the mechanisms underlying podocyte injury, likely providing improved therapeutics for podocyte protection in DN.
The Construction of Major Subject [Grant no (YNZDXK202401)] of Huadu District People's Hospital of Guangzhou;Guangzhou Science and technology plan project(202201011649);The Internal Fund of Huadu District People's Hospital of Guangzhou (2022C04)
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
