
Research Article | DOI: https://doi.org/10.31579/2768-2757/193
North Manchester General Hospital, Department of Urology, Manchester, M8 5RB. United Kingdom.
*Corresponding Author: Anthony Kodzo-Grey Venyo., North Manchester General Hospital, Department of Urology, Manchester, M8 5RB. United Kingdom.
Citation: Grey Venyo AK, (2026), Synovial Sarcoma of The Kidney: An Update, Journal of Clinical Surgery and Research, 7(1); DOI:10.31579/2768-2757/193
Copyright: © 2026, Anthony Kodzo-Grey Venyo. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 15 December 2025 | Accepted: 31 December 2025 | Published: 08 January 2026
Keywords: synovial sarcoma of kidney; renal synovial sarcoma; spindle cell tumour; biopsy; histopathology; immunohistochemistry; molecular and cytogenetics studies; nephrectomy; radiotherapy; chemotherapy; aggressive; poor prognosis; further studies
A Synovial sarcoma (SS) is an uncommon mesenchymal tumour entity which accounts for 5% to 10% of soft tissue sarcomas (STS). Primary renal synovial sarcoma (PRSS) is a very rare, rapidly growing tumour, which is associated with the potential for the development of metastatic disease. The main prognostic factors of PRSS include the size of the tumour, the histopathology grade, as well as translocation t (X; 18) (p11.2; q11.2) (fusion of SYT gene -chromosome 18- with SSX genes (1, 2 or 4)-chromosome X) is recognised as the commonest pathognomonic sign. Aggressive surgical resection of the tumour with complete excision of the together with concomitant regional lymphadenectomy is the treatment of choice for majority of PRSSs, while additional en bloc resection of the adjacent affected organs is often undertaken. At the moment, the role of pre-operative or post-operative chemotherapy has remained equivocal. The prognosis of patients with PRSS has tended to be poor, as the 5-year survival rate has been documented to vary from 20% to 30% in reported cases and studies and further deterioration in the outcome pursuant to treatment has been documented in association with the pathology examination finding of a high mitotic activity. Local recurrence even after complete radical excision of the kidney tumour has remained the most frequent recurrence which eventually emanates in the development of distant metastases and death of patients.
Synovial sarcoma (SS) is stated to be a rare mesenchymal tumour entity which accounts for 5% to 10% of soft tissue sarcomas (STS) [1] [2]. Lejars and Rubens-Duval were stated to have reported and initially named the tumour as synovial endothelioma in 1919. The most predominant postulate for the origin of synovial sarcoma was iterated to related to the retrograde differentiation of an undefined mesenchymal cell [1] [3] [4]. It has been iterated that this type of tumour could be encountered either within the extremities close to articulations in about 85% to 95% of cases or bursas, sinews and the head and neck region in about 10% of cases [1] [5]. It has also been iterated that synovial sarcoma could be identified in very unusual parts of the human body without correlation to the joints, including the nervous system, thoracic and abdominal wall cavity, prostate, fallopian tubes, retroperitoneum, bones and the kidneys [1] [5] [6] [7] [8] [9]. Primary renal synovial sarcoma (PRSS) has been stated to constitute between 1% and 3% among all kidney tumours [1] [10]. It had been iterated that the first description of synovial sarcoma of the kidney was in 1999 by Faria, while it had been previously categorized as an embryonal sarcoma of the kidney [1] [11] [12]. It has been pointed out that the clinical manifesting features of PRSS had typically ranged from presence of an enlarged abdominal mass, vague pain and haematuria to local invasion as well as liver and lung metastatic disease [1] [13] [14] [15] [16] [17]. In view of the limited number reported sporadic cases and case series of PRSSs, there is so far no global consensus opinion guidelines related to the diagnosis and treatment of this uncommon tumour entity. There is therefore a global need for guidelines to be agreed upon regarding the diagnostic criteria and options of treatment for this rare aggressive tumour. [1] [18] [19] [20] [21]. Before guidelines can be made regarding the diagnostic features and management options, it would be necessary for urologists, oncologists, and pharmacotherapy research workers, radiologists and pathologists would need to study the tumour carefully and multi-disciplinary global treatment trials need to be established quickly and clinicians who treat new cases of PRSS should be encouraged to report their cases in the literature so that lessons can be learnt from their experiences. This article on synovial carcinoma of the kidney is divided into two parts (A) Overview which has discussed the general overview aspects of synovial sarcoma and (B) Miscellaneous narrations from some case reports, case series and studies on synovial sarcoma of the kidney.
To review and update the literature on synovial sarcoma of kidney.
Internet data bases were searched including: Google; Google Scholar; Yahoo; and PUBMED. The search words that were used included: Synovial sarcoma of kidney; Renal synovial sarcoma. Eighty-four (84) references were identified which were used to write the article which has been divided into two parts (A): Overview which has discussed general overview aspects of synovial sarcoma and (B) Miscellaneous narrations from some case reports, case series and studies on synovial sarcoma of the kidney.
[A] OVERVIEW [22]
Definition / general statement
Essential features [22]
Terminology
Epidemiology [22]
Sites [22]
With regard to the sites of the human body that could be afflicted by synovial sarcoma, synovial sarcoma can occur anywhere within the body and the primary site distribution has been summated as follows: [23]
Pathophysiology [22]
Aetiology [22]
Clinical features
It has been documented that synovial sarcoma is very rarely associated with previous radiotherapy [29]
Diagnosis [22]
Radiology description
The radiology imaging features of synovial sarcoma had been summated as follows: [22]
Prognostic factors [22]
Treatment
The treatment options of synovial sarcoma had been summated as follows:
Gross description
Summations made on the macroscopy pathology examination features synovial sarcoma include the ensuing: [22]
Microscopic (histologic) description
The ensuing summations had been made regarding the microscopy pathology examination features of synovial sarcoma of the kidney: [22]
Immunohistochemistry staining studies
Positive stains
It has been stated that immunohistochemistry staining studies in synovial sarcoma cases demonstrate that the tumour cells exhibit positive staining for the following tumour markers:
Negative stains
Electron microscopy description
Molecular / cytogenetics description
Differential diagnosis
[B] Miscellaneous Narrations and Discussions from Some Case Sreies and Studies Related to Synovial Sarcoma of Kidney
El Chediak et al. [69] reported a 26-year-old male who had experienced recurrent flank pain and visible haematuria over several months. He had had ultrasound of renal tract which demonstrated a lower pole mass that was concerning for renal cell carcinoma. After confirmation of a right kidney tumour, which measured 6 cm, by a contrast-enhanced CT scan, he underwent right radical nephrectomy with para-caval lymph node dissection, at another institute, with pathology examination of the tumour in the hospital which initially was interpreted as depicting features of as an adult type of Wilm’s tumour. After his referral to the institution of El Chediak et al. [69] the pathology slides of the kidney tumour were re=examined by the pathologist of the new establishment, where the morphology and immunohistochemistry staining profiles were analysed, and results were adjudged to be consistent with the diagnostic features of synovial sarcoma of the right kidney. The tumour was described as monophasic and had shown a cellular spindle cell proliferation with a prominent perivascular growth pattern and partial necrosis. The tumour cells had exhibited positive staining for vimentin, BCL-2, CD56, MCK (partial), and negative staining for CD10, 31, 34, 99, 117, CK7, Desmin, SMA, MyoD1, EMA, WT-1, S100, RCC, PAX8, GATA-3, and Synaptophysin (see figure 1).
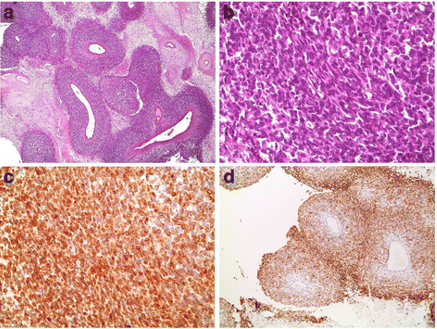
Figure 1: Partially necrotic, densely cellular proliferation with a prominent perivascular growth pattern (a, H&E stain, 40×). Tumor cells are essentially spindle in appearance (b, H&E stain, 400×), and express vimentin (not shown), focal keratin (not shown), BCL-2 (c, 400×), and CD56 (d, 100×) Reproduced from: [69] under creative commons attribution license.
Molecular studies on the paraffin-embedded blocks were undertaken to test for the t(X; 18) SYT/SSX fusion transcript, utilising RT-PCR, at the University of Michigan Health System. RT-PCR amplification was undertaken using fluorescent dye-labelled primers, that are specific for the SYT-SS18 and SYT-SSX genes. The PCR products were then detected and sized by capillary electrophoresis to identify the presence of chimeric transcripts. A concurrent internal control was run to ensure the integrity of the mRNA. FISH analysis was also undertaken utilising a break-apart style probe. The results were unfortunately negative due to the low-quality samples. According to these findings, a diagnosis of primary monophasic SS of the kidney was made. It was elected for serial follow up and no adjuvant treatment, thereafter. Six months subsequently, a follow-up CT scan had identified a 1.5 cm x 1.7 cm left lower lobe lung nodule which was suggestive of metastasis. Consequently, he underwent a smooth left lower lobe wedge resection. Fusion gene product analysis on the resected lung tissue, via FISH, revealed SYT-SSX 2 gene rearrangement had confirmed features of SS and a diagnosis of was confirmed. Three months subsequently, he had CT scan of the chest, abdomen, and pelvis which demonstrated another disease recurrence in the nephrectomy surgical bed, with tumour invasion of the inferior vena cava and the presence of conglomerate suspicious aorto-iliac lymph nodes. A multidisciplinary team meeting discussion was held where it was decided to commence the patient on Doxorubicin 50 g/m2 and Ifosfamide 5 g/m2 chemotherapeutic regimen. Following the third cycle, of combination chemotherapy, he had CT scan and MRI, which demonstrated 30% to 50% interval decrease in size of the tumour masses in the right nephrectomy bed and adjacent retroperitoneum, IVC tumour, and distal aortocaval lymph nodes, which indicated partial treatment response. The patient received a total of 5 cycles, with no adjunct side effects. He had a follow-up MRI scan, several months subsequently, which revealed continued decrease in the size of 3 masses at the previous surgical site, inferior vena cava (IVC) tumour invasion, and aortocaval lymph nodes, indicating continued response to treatment. One of the small masses in the nephrectomy bed had almost completely resolved, upon radiology imaging, with no new progression. It was then decided to have the patient undergo surgical resection of the residual masses at the previous surgical bed with removal of the aorto-caval lymph nodes, thrombectomy with vena cava repair. All surgical margins were negative. The final pathology examination of the conceived tumour demonstrated necrosis, with no viable tumour identified. Thus, a complete pathology response was achieved utilising the Adriamycin/Ifosfamide regimen, a year after the initial nephrectomy. A sample of the kidney lysate was again tested for the (X; 18) SYT/SSX fusion transcript via RT-PCR and FISH, and results were negative, suggestive of complete treatment response.
El Chediak et al. [69] made the ensuing conclusions:
Kim et al. [70] reported two cases of primary synovial sarcoma of the kidney. Both patients had a mass within the upper part of the right kidney without any primary extrarenal neoplastic lesions. Grossly, the tumours were found to be soft to rubbery masses they had measured 5.5 cm and 5 cm in diameter, respectively. Histologically, both tumours were found to be poorly differentiated synovial sarcoma. The lesions had exhibited a hypercellular solid or lobular growth of round, oval, or short spindle cells in variably solid sheets, in intersecting fascicles, or in a haphazard fashion. Areas of solid aggregation or fascicles of the tumour cells alternating with hypocellular myxoid tissues, together with areas displaying a prominent hemangiopericytoma-like pattern, were identified. Immunohistochemistry staining studies of the tumour had demonstrated that the tumour cells had exhibited diffusely positive staining for vimentin, and a few tumour cells had exhibited positive staining for cytokeratin, epithelial membrane antigen, and neurofilament. The tumour cells exhibited negative staining for S-100 protein, CD34, smooth muscle actin, and desmin, whereas CD56 and CD99 were positive. In both cases, reverse transcription–polymerase chain reaction utilising ribonucleic acid extracted from formalin-fixed, paraffin-embedded tissues identified SYT-SSX2 fusion gene transcripts, which were characteristic molecular findings of synovial sarcoma. One patient died 10 months after the initial diagnosis. Kim et al. [71] concluded that these tumours (synovial sarcomas) are unique cases of primary synovial sarcoma of the kidney which were confirmed by molecular study. Divetia et al. [71] stated that the renal parenchyma is a rare site of origin for primary synovial sarcoma (SS). Divetia et al. [71] described the clinicopathology, immunohistochemical, and molecular analysis of 7 cases of synovial sarcoma of the kidney. There were 5 female and 2 male patients, whose ages had ranged between 15 years and 46 years. They had manifested with solitary renal masses which had ranged in size from 10.0 cm to 17.0 cm in greatest dimension. Radical nephrectomy was undertaken in all cases. Upon macroscopy examination, the tumours were noted to be large, partially necrotic, and they were observed to contain smooth-walled cysts in 4 cases. Histologically, the tumours were typified by monomorphic spindle cells with indistinct cell borders arranged in intersecting nodular foci with hypocellular myxoid areas, together with a prominent hemangiopericytomatous pattern. The cysts were lined by hobnailed cells with eosinophilic cytoplasm. Immunohistochemistry staining studies of the tumour had demonstrated that the tumour cells had exhibited positive staining for BCL-2 in all 6 cases in which it was performed, followed by vimentin with positive staining in 4 out of 5 cases and for MIC2 (CD99; positive staining in 2 out of 5 cases, calponin with positive staining in 2 out of 2 cases, and epithelial membrane antigen with positive staining in 1 out of 4 cases. Stains for cytokeratin and CD34 were consistently negative. Reverse transcription–polymerase chain reaction (RT-PCR) utilising RNA extracted from formalin-fixed paraffin-embedded tissues was carried out in 4 cases and SYT-SSX fusion gene transcript, which is the diagnostic hallmark of SS, was detected. Two patients developed pulmonary metastasis and died 6 and 12 months after diagnosis, respectively. Divetia et al. [71] concluded that:
Chen et al. [72] reported a case of primary renal synovial sarcoma (SS) in a 48-year-old man. The patient manifested with haematuria and he was found to have a large tumour within his left kidney upon computed tomography scan. Pathology examination of the tumour specimen demonstrated a highly cellular spindle cell neoplasm with minimal pleomorphism. The major differential diagnoses included leiomyosarcoma, hemangiopericytoma, and synovial sarcoma. The presence of focal areas with a biphasic pattern, uniformly positive on immunohistochemistry staining for bcl-2, focally positive staining for epithelial membrane antigen and cytokeratin, and negative immunohistochemistry staining for CD-34, smooth muscle actin and S-100 established the diagnosis. The diagnosis of synovial sarcoma was subsequently confirmed by molecular testing for t(X;18) translocation. Chen et al. [72] stated the following:
Abbas et al. [73] stated that synovial sarcoma (SS) is a soft tissue, generally deep- seated neoplasms which occurs generally within the proximity of large joints. Abbas et al. [73] reported of a case of a 33-year-old man who was diagnosed with primary SS of the kidney which is an extremely rare tumour that accounts for less than 2% of malignant renal tumours. Abbas et al. [73] made the ensuing iterations:
Schoolmeester et al. [74] reported the clinicopathology and immunohistochemistry staining features of 16 cases of genetically confirmed primary synovial sarcoma of the kidney. The cases had afflicted 9 men and 7 women whose ages had ranged from 17 years to 78 years and their mean age was 46 years. The tumours were grossly large, solid, and variably cystic and they had measured between 2.2 cm to 19.0 cm and their mean measurement was 8.6 cm. Microscopically, all the tumours were found to be the monophasic type and diffusely immunoreactive for TLE1 and BCL-2. Focal pankeratin positivity was found in just under half. Ten cases were reported to have carried an SS18-SSX2 fusion transcript, and 5 cases had shown an SS18-SSX1 transcript by reverse transcription polymerase chain reaction. The remaining case had demonstrated SS18 rearrangement by fluorescence in situ hybridization. Clinical follow-up information was available for 12 patients and the follow-ups had between 1 month to 77 months with a mean follow-up of 32.5 months. Fourteen patients underwent radical nephrectomy, and 3 patients had lung metastases at presentation. Six patients died of disease within 1 month to 58 months (mean, 31 months) of their diagnosis. Five patients were alive without evidence of disease 12 to 77 months (mean, 39 months) after surgery. A single patient was alive with metastases to the spine 11 months after surgery. Schoolmeester et al. [74] made the following conclusions:
Chung et al. [76] stated that primary synovial sarcoma arising from the kidney is extremely rare. Chung et al. [76] reported two cases with primary renal synovial sarcoma. Chung et al. [76] also reported that both cases were initially diagnosed as renal cell carcinoma. The first case was a 30-year-old woman who had manifested with right flank soreness. The patient had ultrasound scan which demonstrated a multiloculated cystic tumour that measured 9 cm × 7 cm. She underwent hand-assisted laparoscopic radical nephrectomy; there was no recurrence during 15 months of her follow-up. The second case was a 49-year-old woman who had manifested with a palpable mass in the left upper quadrant of her abdomen of 1 month's duration. She had computed tomography scan which demonstrated a heterogeneously contrast-enhanced tumour that measured 13 cm × 11 cm at the left retroperitoneum with displacement of the pancreas and the left kidney. Hand-assisted retroperitoneoscopic radical nephrectomy was undertaken. She had no evidence of recurrence after 27 months of follow-up. Pathology examination of the tumours of the two patients demonstrated histopathology and immunochemistry staining features of synovial sarcoma with coexisting spindle and epithelial cells. Chung et al. [76] made the ensuing suggestion:
Dassi et al. [77] reported a 20-year-old female who had presented with a mild left flank pain of one-week duration, with no associated history of haematuria or any other systemic symptoms. Her clinical examination demonstrated a large non-tender lump which had involved her left lumbar and left hypochondriac region. She had computed tomography scan of her abdomen and pelvis which demonstrated large heterogeneously contrast-enhancing mass of 14.3 cm × 9.4 cm × 8.5 cm over the middle-region and lower pole of her left kidney with areas of necrosis within it. An iso-to-hypodense heterogeneously enhancing thrombus was noted in her left renal vein and adjacent portion of the inferior vena cava [see figures 2 and 3].

Figure 2: CT image showing tumor invading almost whole of kidney and involving IVC. Reproduced from [77] under Creative Commons Attribution License.

Figure 3: CT image (coronal) Reproduced from [77] under the Creative Commons Attribution License.

Figure 4: Macroscopically, tumour seen replacing whole of kidney with tumour thrombus in left renal vein. Reproduced from: [77] under the Creative Commons Attribution License.

Figure 5: Tumour composed of spindle cells arranged in intersecting fascicles alternating with hypocellular areas. Reproduced from: [77] under the Creative Commons Attribution License.

Figure 6: Microscopic image showing spindle cells. Reproduced from: [77] under the Creative Commons Attribution License.
Renal cell carcinoma was the provisional suspected pre-operative diagnosis. Intraoperatively, a tumour mass was visualised which had replaced the whole kidney. Left radical nephrectomy was undertaken and left renal vein ligated flush with the IVC after milking the thrombus into the left renal vein. Gross examination of the specimen demonstrated a tumour that measured 12.8 cm × 11cm × 4.5 cm, and which had replaced the entire renal parenchyma, and which had involved the pelvi-calyceal system and medulla with thin rim of cortex seen all around. Tumour thrombus was visualized within the lumen of left renal vein [4]. Microscopy examination of the tumour had demonstrated that the tumour was composed of spindle cells that were arranged in intersecting fascicles, alternating with hypocellular areas, suggestive of monophasic synovial sarcoma (SS) [see figures 5 and 6]. On immunohistochemistry staining studies, the tumour cells were noted to have exhibited positive expression for: bcl-2, calponin, and EMA. Both Mic-2 and CK were focally positive. Molecular analysis had revealed a translocation between the SYT gene on chromosome 18 and SSX on chromosome X, which was consistent with the diagnosis of synovial sarcoma (SS) of kidney.
Dassi et al. [77] made the following conclusions:
Modi et al. [78] reported a Forty-one-year-old male patient who had presented with pain in his left lumbar region and visible haematuria for 1 month. His past and family history is unremarkable. He had been a chronic tobacco chewer for 10 years and a non-alcoholic. He was referred to the cancer centre based upon the findings in his ultrasound scan of a left renal mass. Upon examination he was found to have a normal height, weight, and body mass index for his age. His vital signs were normal and his performance score by ECOG (eastern cooperative oncology group) was 1. Clinically a non-tender palpable mass was felt over his left lumber fossa of around 5 cm × 5 cm with smooth surface and hard consistency. Pallor was present in his sclera and no lymphadenopathy or icterus was identified. He had a CT scan which demonstrated an enlarged left kidney which had almost been completely replaced with heterogeneously hypodense material. There was a hypodense filling defect noted in his left renal vein which had extended up to inferior vena cava suggestive of tumour thrombosis (see figure 7). The results of his laboratory blood test investigations were normal except haemoglobin of 6.7 gm%, serum creatinine of 2.1 mg/dL, and serum BUN of 25 mg/dL.

Figure 7: CT image shows enlarged left kidney and it is almost completely replaced with heterogeneously hypodense material Reproduced from: [78] under the Creative Commons Attribution License.

Figure 8: Lower power view shows round to spindle cells with hemangiopericytoma pattern with areas of hyalinization in between. Reproduced from: [78] under the Creative Commons Attribution License.

Figure 9: High power view shows entrapped normal renal tubules. Reproduced from: [78] under the Creative Commons Attribution License.

Figure 10: The figure shows CD99 positivity. Reproduced from: [78] under the Creative Commons Attribution License.

Figure 11: The figure shows BCL2 positivity. Reproduced from: [78] under the Creative Commons Attribution License.
Histopathology examination of the biopsy specimen from the left renal mass demonstrated round to spindle cells with hemangiopericytoma pattern and area of hyalinization (see figure 8). High power view showed entrapped normal renal tubules (see figure 9). Immunohistochemistry (IHC) staining of the biopsy specimen showed that the tumour cells had exhibited positive staining for CD99 (see figure 10), BCL2 (see figure 11), and vimentin and negative staining for AE1, epithelial membrane antigen (EMA), and leukocyte common antigen (LCA). According to morphological and IHC findings final diagnosis of primary renal synovial sarcoma was made. The patient was found to be clinically inoperable upfront according to Urooncology surgeon. In view of this, he was subsequently offered palliative chemotherapy in form of ifosfamide and adriamycin. He had a CT scan of abdomen which demonstrated partial response after 3 cycles of chemotherapy according to RECIST criteria. Modi et al. [78] made the ensuing conclusions:
Tranesh et al. [79] made the ensuing conclusions:
None.
Acknowledgements to:
Case Reports in Pathology and Hindawi Publishing Limited for granting permission for reproduction of figures and contents of their Journal article under copy right: Copyright © 2014 Gaurang Modi et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
