
Research Article | DOI: https://doi.org/10.31579/2693-7247/116
1 Department of Pharmaceutical and Medicinal Chemistry, Faculty of Pharmaceutical Sciences Ahmadu Bello University, Nigeria.
2 Department of Chemistry and Biochemistry, The University of Texas at Dallas, USA.
3 Department of Chemistry, Chrisland University, Nigeria.
4 Department of Material Science and Engineering, University of Florida, USA.
5 Department of Chemistry, Middle Tennessee State University, USA.
6 Faculty of Pharmacy, Ahfad University for Women, Sudan.
*Corresponding Author: Sodeeq Babalola, Department of Pharmaceutical and Medicinal Chemistry, Faculty of Pharmaceutical Sciences Ahmadu Bello University, Nigeria.
Citation: Sodeeq Babalola, Nosakhare Igie, Isaiah Odeyemi, Abdullahi Y. Idris and Sanni Y. M et al, (2023). Synergistic Drug-Drug Interactions (Ddis) Effects on Anti-Inflammatory Activities Of N-Phenyl-2,4-Dichlorophenyl Hydrazone and Piroxicam. J. Pharmaceutics and Pharmacology Research, 6(1); DOI:10.31579/2693-7247/116
Copyright: © 2023, Sodeeq Babalola. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 15 December 2022 | Accepted: 16 January 2023 | Published: 01 February 2023
Keywords: drug-drug interactions; pharmacokinetic enhancer; inflammation; piroxicam; n-phenyl-2; 4-dichlorophenyl hydrazone
Drug-drug interactions (DDIs) has become an emerging and powerful strategy in drug designing and optimization. In recent years, there has been an increase in research seeking to increase the therapeutic index of already marketed drugs with new chemical entity serving as actual activity enhancers as well as synergistic relationships. Herein, we report the synergistic drug-drug interactions of compound 1-(2,4-dichloro benzylidene)-2-phenyl hydrazine with piroxicam as an enhancer of efficacy showcasing faster onset of action with 48.09% inhibition and longer duration of action with 86.48% inhibition. This is compared to piroxicam’s moderate onset of action with 35.50% inhibition and shorter duration of action with 60.25% inhibition. Compound 1-(2,4-dichloro benzylidene)-2-phenyl hydrazine has also demonstrated potential increase in therapeutic index as a combination therapy with superior efficacy to NSAIDs-proton pump inhibitor combination therapy for the treatment of inflammatory diseases.
Drug-drug interactions (DDIs) is the term that covers the results of combining two or more medications, including both desired and undesired consequences. Drug-drug interaction (DDI) studies are intended to identify how one drug may affect another's metabolism and, vice versa. The results of DDI studies always fall into one of these two categories: (a) when a second drug increases the first drug's catabolism, decreasing the first drug's efficacy; or (b) when a second drug decreases the first drug's catabolism, increasing the first drug's efficacy (or increasing its toxicity) [1]. Drug interactions are occasionally considered to be detrimental and leading to adverse outcomes. However, in some circumstances, some are involved in the enhancement of treatments through combinatorial approach. This action is accomplished through; targeting an identified mechanism of resistance, inhibiting a metabolic pathway to enhance the pharmacokinetic (PK) profile of the primary drug, and enhancing a pharmacological activity. Examples include levodopa (L-Dopa) and dopadecarboxylase inhibitors, ꞵ-lactam antibiotics and clavulanic acid, 5-fluorouracil (5-FU) and folinic acid, and penicillin and probenecid.
Recently, kenpaullone, an inhibitor of glycogen synthase kinase GSK3β has been identified to enhance the activity of the alkylating agent, temozolomide in glioblastoma. This was achieved by increasing the apoptosis-inducing activity of temozolomide through attenuation of stem-cell properties of glioblastoma cells [2,3]. Another example is the enhancement of paclitaxel activity in tumoral cells by Carba1, a microtubule-destabilizing carbazole derivative. Although paclitaxel stabilizes the microtubules, Carba1's modification of tubulin dynamics encouraged paclitaxel accumulation in the microtubules, leading to increased efficacy [2,4].
Pharmacodynamic interactions, also known as pharmacodynamic DDIs, are DDIs that result from the combined pharmacodynamic actions of the interacting drugs [5]. The second drug is often chosen based on one of these factors: (a) Both drugs are used to treat the same condition. This is the case with the DDI trial involving the study drug regorafenib and the second drug, irinotecan. When treating colorectal cancer, irinotecan and regorafenib are frequently co-administered. (b) The second drug is for a separate condition, given at the same time as the study drug, such as a contraceptive, statin, acetaminophen, warfarin, or a monoamine oxidase inhibitor. The condition that the second drug is intended to treat in this instance is different from that of the study drug [1].
Pharmacokinetic studies often involve a second drug. This relates to clinical pharmacokinetic studies that are used to predict which classes of drugs are most likely to participate in DDIs with the study drug rather than to assess effectiveness or safety. In this case, the second drug is a recognized inhibitor of a cytochrome P450 enzyme and is typically employed as a model "second drug" in research on drug-drug inhibition [1]. Example is the employment of ketoconazole in clinical investigations of DDIs as a model "second drug." Ketoconazole inhibits cytochrome P450 3A4 when utilized in research on drug-drug interactions (CYP3A4). Although the medicine ketoconazole is technically an antifungal, this sort of clinical pharmacokinetic investigation that employs ketoconazole has no relation to the treatment of fungi [1]. Sometimes the association of an already marketed drug with typically one adjuvant molecule improves both the activity and potency of the drug. More recently, some drug-drug combinations have been included in a modern drug design strategy. These small molecules, also known as enhancers, boosters, or activators, hardly ever or never directly affect health. They can, however, enhance the action of a medicinal drug already in use, leading to greater therapeutic effects [2].
Pharmacokinetic (PK) interactions (PK DDIs) sometimes called dispositional interactions is the modification of one drug's PK or disposition (ADME) by another, which is the hallmark of these interactions. If these changes are severe enough, the exposure to the affected drug may change significantly, eventually modifying how the drug works [6]. In terms of PK, exposure is measured by the region enclosed by a drug's plasma concentration versus time curve. The area under the curve, or AUC, is used to describe that region. The pharmacological benefits which may be enhanced efficacy and/or undesirable side effects may manifest if the exposure (i.e., AUC) is greatly elevated. The therapeutic effects might not manifest if the exposure is sufficiently reduced, and therapeutic failure can result. Changes in the peak plasma concentration (Cmax) of one drug brought on by another drug may occasionally be sufficient to have clinically relevant adverse effects [6].
The "victim" drug in a DDI is the drug whose disposition is altered, and the "perpetrator" drug is the one that alters the victim's disposition [6]. A perpetrator might be expected to increase the AUC of a victim drug by slowing its metabolism in the liver or the intestine, blocking the efflux of the victim drug from hepatocytes into the bile, or increasing its oral absorption. This could happen if the perpetrator decreased protein or tissue binding of the victim drug, reduced its renal secretion into the urine, inhibited the transporter-mediated efflux of the victim drug back from intestinal epithelial cells into the lumen of the intestine, or slowed its intestinal metabolism [6].
On the other hand, a perpetrator may be anticipated to reduce the AUC of a victim drug by limiting its absorption or bioavailability, speeding up its metabolism in the liver or intestine, or by increasing its efflux from intestinal cells into the intestine's lumen or from hepatocytes into bile. Theoretically, a perpetrator drug's involvement with any aspect of the ADME of a victim drug might change the victim drug's AUC. This broad mechanistic view of DDIs allowed for the long-term prediction of several two-drug DDIs [5]. The vast majority of metabolic DDIs involve the inhibitors, inducers, and substrates of the cytochrome P450 enzymes (CYPs). Although, theoretically any inhibitor or inducer of any drug-metabolizing enzyme could act as a perpetrator of DDIs and any substrate of any drug-metabolizing enzyme could become a victim. This is largely because CYPs predominate over all other types of enzymes in the metabolism of drugs [5].
Elvitegravir is a dramatic and unusual case since it is one of the few times when the package label calls for simultaneous administration of a CYP enzyme inhibitor. The CYP enzyme inhibitor was used to boost the study drug's exposure. Another pharmacokinetic enhancer, cobicistat, is used to inhibit CYP3A and stop the degradation of certain drugs by CYP3A. For inhibiting CYP3A-mediated drug catabolism, ritonavir or cobicistat are referred to as helpful "pharmacokinetic boosters or enhancers" [1].
P-gp controls how well pharmaceuticals are absorbed through the intestines, therefore medicine that can stimulate or inhibit P-gp activity can lead to the development of DDI. The bioavailability of drugs that are ineffectively absorbed can be greatly increased by P-gp inhibition. It is known that sildenafil decreases Pgp's transporter function, providing a potential method to increase the availability and perhaps even the effectiveness of anticancer drugs [7].
Drug interactions that affect distribution may be caused by interactions with drug transporters and drug displacement from plasma proteins, most notably serum albumin [8]. Displacement of one drug or substance by another drug or substance from its binding site or sites, which results in a rise in the concentration of the free drug, is an example of a drug interaction of widespread clinical concern but unusual clinical importance. Higher distribution and clearance take place as a result of the increased free drug concentration, and the pharmacodynamic action is also improved [8]. In this situation, drugs that have a high affinity for plasma proteins, a small volume of distribution (Vd 1 L/kg), a prolonged half-life, and a low therapeutic index might have negative clinical consequences.
The pharmacological displacement seen when warfarin and diclofenac are co-administered is a classic example. Because diclofenac and warfarin have a similar affinity for albumin, when it is given to a patient who is taking warfarin on a long-term basis, the warfarin is dislodged from its binding site. Serious hemorrhagic responses emerge as free warfarin concentrations in the plasma rise [9].
The drug's elimination from the body might experience a variety of interactions in the organ from which it is expelled [10]. When two or more medications employ the same transport route, DDI may result from a process of competition at the level of active tubular secretion. One example is how NSAIDs affect the amount of methotrexate toxicity when the anti-proliferative drug's renal excretion is inhibited [11].
The interactions between the drugs can, however, be used therapeutically. Probenecid, for example, can raise the serum levels of cephalosporins and penicillins, delaying their renal elimination and reducing dose requirements. Probenecid works by interfering with an organic anion transporter in the renal tubules by competitive inhibition, which raises the plasma concentrations of other transporter substrates while decreasing excretion [12].
Inflammation is a biological defense mechanism that occurs when the body's tissue homeostasis is disrupted by the presence of biological, chemical, or physical agents. The immune system produces a number of pro-inflammatory mediators, but when these mediators are overproduced, as it happens in chronic inflammation, it can result in the development of a number of chronic diseases. As a result, it becomes crucial to slow down the inflammation process, and for this reason, non-steroid anti-inflammatory medicines are typically employed with the risk of negative side effects [13].
Non-steroidal anti-inflammatory drugs (NSAIDs) have become widely used as analgesics, antipyretics, and anti-inflammatory therapies across the world. The market size for NSAIDs in the United States was estimated at $5.78 billion in 2021, and it was expected to reach $7.10 billion by 2025. In 2019, the worldwide NSAIDs market was worth $15.58 billion, with a forecast of $24.35 billion by 2027 [14,15]. One of the major reasons driving the NSAIDs market is the rising prevalence of pain and inflammation-causing disorders [14]. In this study, we examine the anti-inflammatory activity of compound N-phenyl-2,4-dichlorophenyl hydrazone and it effect as efficacy enhancer in inflammation therapy.
2.1 Chemistry
All the reagents used were purchased from Sigma Aldrich, and they were used with no further purification. Electrothermal Engineering LTD 9100 apparatus was employed in the melting points determination of the synthesized compounds. The FTIR spectra were recorded on Agilent technologies spectrometer model 543, and the 1H and 13C NMR spectra were obtained using a Brucker AMX 400 MHz spectrometer operating at 400 MHz and 101 MHz respectively with dimethyl sulfoxide (DMSO) used as the solvent. Chemical shifts (δ) are reported in parts per million and are referenced to the NMR solvent peak.
2.1.1 In-silico pharmacokinetics (ADME) and toxicity studies
The drug-likeness studies, in-silico pharmacokinetics, and toxicity studies were evaluated on ADMETlab 2.0 (https://admetmesh.scbdd.com) and Protox-II web (https://tox-new.charite.de). The ADMETlab 2.0 was used to evaluate detailed parameters of drug-likeness, absorption, distribution, metabolism, and excretion. The toxicity studies were conducted on Protox-II and ADMETlab 2.0 web servers.
2.1.2 Synthesis
Equimolar quantities of 2,4-dichloro benzaldehyde (20mmol) and phenylhydrazine (20mmol) were mixed in 30ml of ethanol at room temperature. The mixture was continuously stirred for 3hrs and the progress of the reaction was monitored by TLC. The white crystalline solid formed was filtered off, dried, and then recrystallized from pet ether.
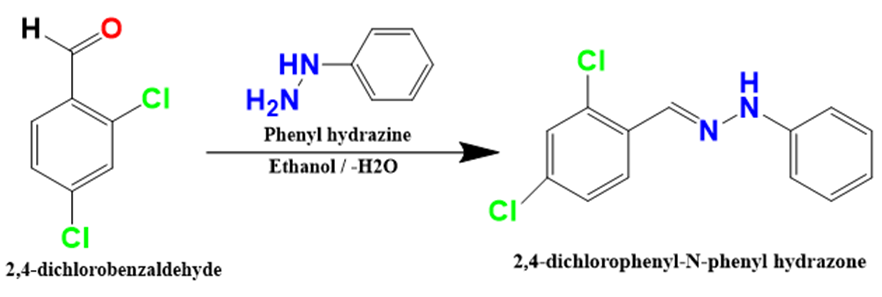
Scheme 1: Synthesis of 1-(2,4-dichloro benzylidene)-2,4-phenyl hydrazine.
2.2 Pharmacology Study
2.2.1 Experimental Animals.
Twenty-eight Swiss albino mice (15-34 g) were used in the experiments. The animals were procured within the Zaria community, Kaduna State, Nigeria. The mice were housed in single-sex cages under a 12-hour light:12 hour dark cycle (lights on at 6 am) in a controlled-temperature room (22 ± 2 °C with 50 ± 10% humidity) at the animal house. The mice were kept for 2 weeks to stabilize, habituate (acclimatize) and become more adult. After 7 weeks, all the animals were adults at the age of 7 weeks and weights of 15–34 g. Availability of standard diet and water was ad libitum. All animal experiments were performed as per the requirement of the bio-ethical committee protocols of the ABU Committee on Animal Use and Care in compliance with the guidelines for the care and use of laboratory animals provided by the National Institute of Health (NIH publication no. 85–23, revised 1985). Ethical approval was sought and obtained from ABU Committee on Animal Use and Care.
2.2.2 Oral Acute Toxicity Studies and lethality (LD50) test.
The acute toxicity and lethality (LD50) of the titled compounds were estimated in mice using the Fixed Dose Procedure (FDP) — OECD TG 420. This OECD guideline method does not use the death of animals as a clear sign of toxicity. Instead, any changes such as changes in skin and fur, eyes and mucous membranes, and also respiratory, circulatory, autonomic, and central nervous systems, somatomotor activity, and behavior patterns. Attentions are particularly directed at observations of increased motor activity, anesthesia, tremors, arching and rolling, clonic convulsions, tonic extension, lacrimation, Straub reaction, salivation, muscle spasm, writhing, hyperesthesia, loss of righting reflex, depression, ataxia, stimulation, sedation, blanching, hypnosis, cyanosis and analgesia, diarrhea, lethargy, sleep, and coma were taken as a sign of toxicity (Deora et al., 2010) [16].
The limit test was employed in the experiment, a group (n = 3) was used for the test compound. The test animals were male Swiss albino mice with weights ranging between 20 – 25 g. The test dose of the titled compound was prepared by dissolving 150mg of each titled compound in 3 ml of deionized water to form an aqueous suspension of 50mg/ml concentration which is equivalent to 2000mg/kg per body weight for mice weighing 25g. The compound was administered orally in the form of deionized water suspension (1% w/v) at an appropriate volume equivalent to a dose of 2000 mg/kg (n = 3). Animals were observed continuously for the first one hour for any toxic symptoms after administration and then for the next 24 hours, 48 hours, 7 days, and 14 days.
2.2.3 Anti-inflammatory activity evaluation.
The anti-inflammatory activity of the titled compound was evaluated using Carrageenan-induced hind paw edema in mice model. The method of Kasahara et al., 1985 [17] as described in our previous study [14].
The measurement of the hind paw was carried out using a thickness gauge vernier caliper before each treatment (Vo) and in each interval (Vt) after the administration of the test compound, dosing vehicle, and the reference drug. The data were expressed as mean ± standard error of the mean (n=5). The data were analyzed statistically using a Two-way analysis of variance (ANOVA) with replication, residual error test, and Tukey’s Multiple Comparison Test. The percentage of swelling inhibition was calculated using the following equation:
Inhibition (%) = {[(Vt − Vo)control − (Vt − Vo)treated]/(Vt − Vo)control} ×100Eq. (4)
Where Vt and Vo relate to the average volume in the hind paw of the mice after carrageenan injection and before carrageenan injection respectively. All the results were expressed as Mean ± Standard Error of Mean (S.E.M.) and percentage of inhibition.
3.1 In-silico pharmacokinetics (ADME) and toxicity studies

Table 1: Absorption
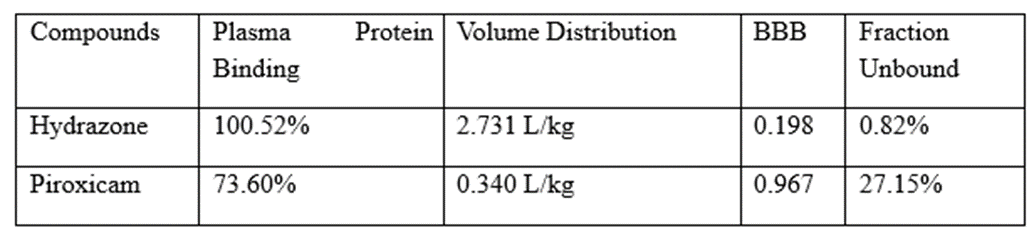
Table 2: Distribution
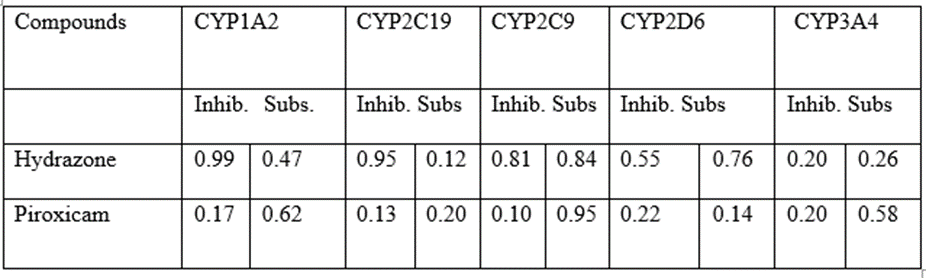
Table 3: Metabolism.
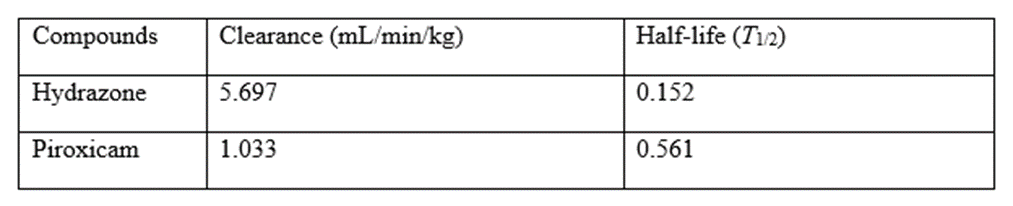
Table 4: Excretion
3.2 Synthesis
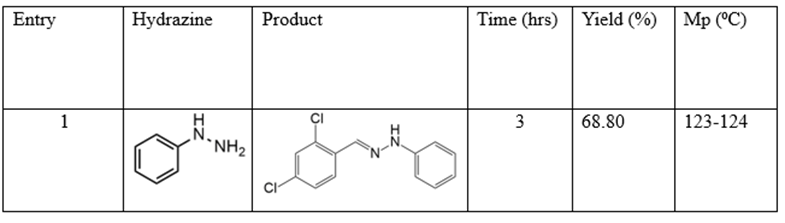
-(2,4-dichlorobenzylidene)-2-phenylhydrazine. Yield 68.80%, Crystalline white solid, mp 123-124 ⁰C. FTIR (KBr, cm-1): 3302 (N-H), 3030 (C-Himine), 1572 (C=N), 1517 (C=Caromatic), 1252 (C-N), 1047 (C-Cl). H1 NMR spectrum (400 MHz, DMSO-d6) δ, ppm: H1 NMR spectrum (400 MHz, DMSO-d6) δ, ppm: 7.01 d (1Harom, J = 7.1 Hz), 7.16 d (2Harom, 8.2 Hz), 7.22 d (2Harom, 7.8 Hz), 7.47 (Harom, 8.5 Hz), 7.63 (Harom, 1.7 Hz), 8.09 (Harom, 8.4), 8.19 (Himine), 11.23 (1H, NH). 13C NMR spectrum (101 MHz, DMSO-d6), δ, ppm: 112.30, 126.11, 126.54, 127.94, 128.62, 129.56, 131.89, 133.18, 134.34, 136.91, 139.89.
Table 5: Synthesis of N-phenyl-2,4-dichlorophenyl hydrazone.
3.3 Pharmacology
3.3.1 Oral Acute Toxicity Studies and Lethality

Table 6: Oral Acute Toxicity Studies and Lethality (LD50) results.
3.3.2 Anti-inflammatory activity evaluation results.
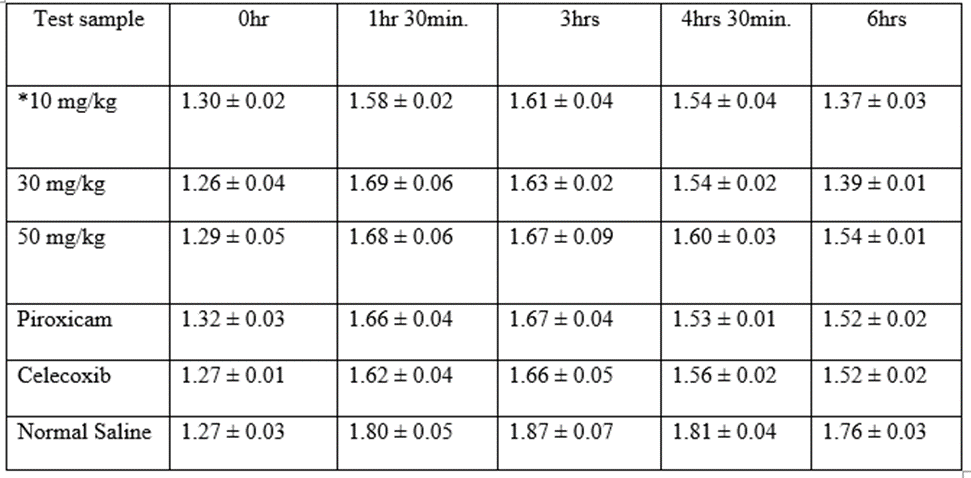
* = Co-administration of 10 mg/kg each of the hydrazone and piroxicam. Mean ± S.E.M of hind paw thickness, n=5, p-value = 0.00064 < α = 0.05, F = 5.31 > F Critical = 2.46
Table 7: Mean ± S.E.M of hind paw thickness at 10mg/kg dose. n=5
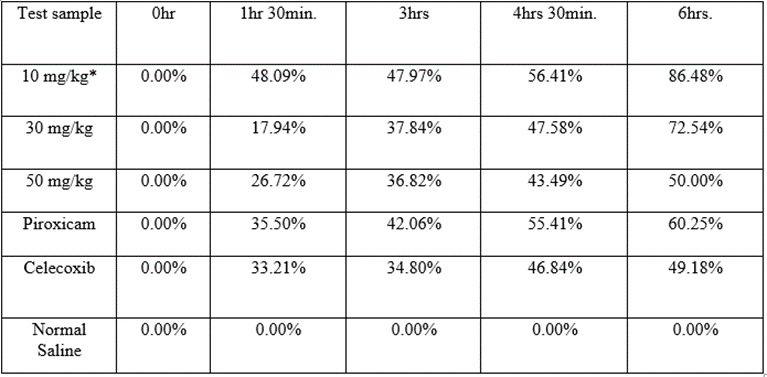
* = Co-administration of 10 mg/kg each of the hydrazone and piroxicam.
Table 8: Percentage inhibitions of hind paw edema at 10mg/kg dose.
4.1 Chemistry
Twenty minutes after the start of the reaction white crystalline solids were produced with 68.80% yield after three hours according to results in Table 5. The melting point was deduced to be 123-124 ⁰C. Spectroscopic analysis of the purified product indicated N-H stretching, imine C-H
stretching, and C=N stretching are features of the FTIR absorption signals at 3302 cm-1, 3030 cm-1, and 1572 cm-1. The hydrazone functional group's synthesis in the molecule was verified by the singlet proton peaks' locations at 8.19 ppm and 11.23 ppm as well as by the C-13 peak's location at 136.91 ppm [18].
4.2 Pharmacology
4.2.1 Oral acute toxicity and lethality studies
The oral acute toxicity studies and LD50 of compound 1-(2,4-dichloro benzylidene)-2-phenyl hydrazine was evaluated at 2000 mg/kg dose in mice according to OECD guidelines. There was observed motor activity reduction and slight sleepiness which summed up as lethargy as a sign of toxicity within the first hour of administration. This symptom wears off after the first our oral administration with no further sign of toxicity over the fourteen days of study. This examination implies a remote risk of acute intoxication and indicates a high degree of relative safety for the oral administration of these compounds according to results in Table 6. Furthermore, there was no mice death count observed during the fourteen days study, implying that the lethality dose of compound N-phenyl-2,4-dichlorophenyl hydrazone is greater than 2000 mg/kg dose. By this, the LD50 of the compound 1-(2,4-dichloro phenyl)-2-phenyl hydrazine can be said to fall into Class V of the globally harmonized system (GHS) (2000 mg/kg < LD50>
4.2.2 Anti-inflammatory activity
Compound 1-(2,4-dichloro benzylidene)-2-phenyl hydrazine demonstrated statistically significant (p-value < 0>Table 7.
The test compound was observed to exhibit slow onset of action at 30 mg/kg and 50 mg/kg doses with percentage inhibitions of 17.94% and 26.72
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
