
Research Article | DOI: https://doi.org/10.31579/2690-1897/209
1Department of Medicinal Chemistry, Faculty of Pharmacy, University of Tripoli, Tripoli, Libya.
2Department of Chemistry and Toxins, Judicial Expertise and Research Center, Tripoli, Libya.
3Department of Biosciences, University of Salzburg, Salzburg, Austria.
4Zoology Department, Faculty of Science, Tripoli University, Tripoli, Libya.
*Corresponding Author: Abdul M Gbaj, Professor of Genetics and Biochemistry, Department of Medicinal Chemistry, Faculty of Pharmacy, University of Tripoli, Libya.
Citation: Nisreen H Meiqal, Sofian S. Mohamed, Shahrazad Ali Eteer, Jamal Ali M Elbakay, Inass A Sadawe, et al, (2024), Symmetrical 1,2-Phenylenediamine Schiff’s Base Derivatives as New Potential Dna Intercalators, J, Surgical Case Reports and Images 7(7); DOI: 10.31579/2690-1897/209
Copyright: © 2024, Abdul M Gbaj. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 02 August 2024 | Accepted: 13 August 2024 | Published: 20 August 2024
Keywords: schiff’s base; dna intercalator; anticancer; ortho-phenylenediamine; molecular docking
Fifteen symmetrical 1,2-phenylenediamine Schiff’s base derivatives were designed as DNA intercalators. Adsorption, distribution, metabolism, elimination, and toxicity (ADMET) properties and drug-likeliness of the designed compounds were predicted by Swiss-ADME software, and the molecular docking study was performed in the PyRx tool. Four Compounds NHM01, NHM04, NHM06 and NMH11 were synthesized and the structure of each compound was analyzed using Fourier transform infrared spectroscopy (FTIR), 1H NMRand13C NMR. Moreover, the four compounds were in vitro biologically screened for their interactions with the genomic DNA. The binding properties of these compounds to genomic DNA (G-DNA) were investigated by UV-visible absorption and fluorescence spectroscopy. The molecular docking studies of compounds revealed that all the compounds are bioactive, however, compound NHM01 shows significant DNA binding affinity over standard drugs. The biological results indicate that the four synthesized compounds can interact with G-DNA by intercalation binding. Compound NHM01 showed the highest key selection vector (KSV) value, followed by NHM04, which suggests that compound NHM01 binds most tightly to G-DNA. Our results demonstrate that NHM01 and NHM04may serve as novel lead compounds for the discovery of more 1,2-phenylenediamine Schiff’s base derivatives with improved anticancer potency and selectivity.
Deoxyribonucleic acid (DNA) is the storage unit of genetic information and considered as major biological target for numerous anticancer agents. The anticancer agents that target DNA generally interact with DNA through reversible binding by three main modes: static electronic interactions, groove binding, or intercalation. The static electronic interactions (external surface binding) could be related to the cationic molecules that bind non-specifically with the anionic sugar phosphate backbone of DNA through outer edge stacking (Serec et al., 2016). The second mode of interaction is groove binding which comprises the insertion of the molecule into the base edges of the minor or major groove (usually G•C sites in the major groove, A•T sites in the minor groove).The major groove has multiple sites of interaction, its dimensions are 8.5Å in depth and 11.6Å in width, where, bulky molecules can easily access the major groove. Conversely, the minor groove, which has a depth of 8.2 Å, is narrower and has fewer binding sites, however, binding to the minor groove is the most common. The third mode of interaction is intercalation which is closely related to most anticancer drugs which are currently in clinical use. In essence, small molecules can interact with DNA involving a single or mixed binding mode(Bhaduri et al., 2018). In this paper, our focus will be on DNA intercalation. DNA intercalators area class of compounds that can reversibly bind to DNA through insertion between adjacent base pairs perpendicularly to the axis of the helix. These interactions canlead to conformational changes involving an increase in the vertical separation between base pairs, unwinding and lengthening of the DNA helix. It may also interfere with the detection and function of associated proteins or enzymes leading to the failure of DNA repair systems, transcription processes or replication of the DNA (Biebricher et al., 2015). Numerous clinical intercalating agents are available as anticanceragents, for example daunomycin, doxorubicin, mitoxantrone or dactinomycin. Ethidium bromide is a phenanthridine monomer dye frequently used as a fluorescent marker (nucleic acid stain) in molecular biology for many procedures such as visualization of nucleic acid. Ethidium bromide intercalates between adjacent base pairs in the double-stranded DNA molecule and leads to deformation of the DNA. The deformation of DNA will possibly affect DNA biological processes, such as DNA transcription or replication (Binaschi et al., 2001). DNA intercalators have three main structural features: i) Chromophores, which are planar polyaromatic rings that bind to DNA, ii) cationic moieties, such as protonated amino groups which may interact with the phosphate backbone and iii) side chains that can interact with DNA minor grooves. In general, DNA intercalators are classified into two major classes: monofunctional and bifunctional (El-Adl et al., 2020).Monofunctional intercalators also referred to as simple intercalators. They consist of one intercalating unit, often carrying a positive charge on their ring system (Wadler et al., 1994).Secondly, bifunctional intercalators, also called bis-intercalators orthreading intercalation, have two typically cationic intercalating units separated by flexible linker groups being long enough to permit double intercalation. Since bis-intercalators have two different intercalating moieties, they can cause stronger binding with DNA than simple mono-intercalators (Wadler et al., 1994). Schiff bases are ranked among the most privileged synthetic organic intermediates extensively employed in industrial, pharmaceutical, and medicalconcerns. These compounds are also known as imines or azomethines with a general formula R-CH=N-R’, where R and R’ are an alkyl or aryl group(Peng et al., 2014). Schiff bases have been reported to exhibit a wide spectrum of biological properties and can also interactwith other ligands like ortho-phenylenediamines which leads to diverse and multipurpose applications(Koll, 2003).Bis-Schiff bases are usually synthesized from the condensation reaction of diamines with two equivalents of aldehyde or ketone in 1:2 molar ratio. The bases can be either symmetric or asymmetric structures, depending on the reacting aldehydes or ketones. (Vernekar & Sawant, 2023). Literature review revealed a great interest on ortho-phenylenediamines Schiff complexes and their applications. In contrast, less attention has been focused on the uncoordinated Schiff bases of ortho-phenylenediamines. In this regard, the search for other uncoordinated anticancer agents with improved safety profiles has become an attractive area of study for cancer management(Lucaciu et al., 2022). The main aimof this work is to design and synthesize some symmetrical Schiff’s base derivatives of o-phenylenediamine and study their DNA intercalating ability using an ethidium bromide competition fluorescent assay and UV-visible spectroscopic method. In addition, there is increasing interest to explore how different structural features of 1, 2-Phenylenediamine Schiff’s base derivatives can affect the DNA binding capability using molecular modeling tools, which may serve as a basis for understanding the molecular mechanism of action of the 1, 2-Phenylenediamine Schiff’s base derivatives and can help to design new chemotherapeutic molecules.
2.1Molecular docking
Molecular docking is an in silico method for recognizing the correct binding pose of a DNA-ligand complex and evaluating its strength using various scoring functions for selecting the best pose generated by each molecule to a rank-order. The molecular docking studies of all compounds with DNA were performed using PyRx software to generate grid, calculate dock scores and evaluate conformers. The steps of molecular docking include DNA structure optimization, in silico preparation for standard and designed compounds, and process of docking calculations (Adelusi et al., 2022).
2.1.1 Opimization of DNA molecule
The crystal structures of B-DNA fragments (PDB ID: 1BNA,1D60, 1ZEW, 2DES and 1DCO) were downloaded from the protein data bank(http://www.rcsb.org/pdb), and analyzed for its active site by Biovia Discovery Studio Visualizer (http://accelrys.com). Auto Dock 4.2 software was used to correct the imported DNA coordinates (The corrections involved deleting unwanted water molecules, adding hydrogen atoms (polar only) followed by adding kolloman charge, and saved as pdpqt format used as input in PyRx software.
2.1.2 Designing and in silico preparation of NHM compounds
The starting geometry of all investigated compounds was constructed using Chem3D Ultra (version 12.0, Cambridge soft Com., USA). The optimized geometry of tested compounds with the lowest energy was used in further processes of molecular docking. Discovery studio visualizer was used to all tested compounds in which the compounds were converted to PDB files that will be used as input in PyRx software.
2.1.3 Docking process and analysis
B-DNA crystal structures (PDB ID: 1BNA, 1D60, 1ZEW, 2DES and 1DCO) were loaded in PyRx virtual screening tools as pdbqt format. Standard drug and designed compounds pdb files were loaded and automatically converted to pdbqt format. Docking was performed using Autodock Vina incorporated in Pyrx software. The centers of the grid box were assigned automatically and the dimensions of the box were set to 25 × 25 × 25 Å. After docking, the complexes with the lowest binding energy (kcal/mol) were selected and their interaction modes were visualized by Discovery Studio.
2.2 Prediction of ADMET properties and drug-likeliness
The in silico procedure has become a very important tool in drug identification and screening. Ligand absorption, distribution, metabolism and excretion (ADME) pharmacokinetic properties of the designed Schiff’s base derivatives of o-phenylenediamine, are evaluated to determine their activity within the human body and prediction of these properties in advance will save the cost of drug discovery substantially. To verify drug-likeliness, canonical SMILES format of the designed NHM compounds were used in swissADME (http://www.swissadme.ch/index.php). The main bioavailability parameters were molecular weight (g/mol), lipophilicity (XlogP3), solubility (log S), polarity (Topological Polar Surface Area - TPSA in Å2), saturation (fraction of carbon atoms in Sp3 hybridization – Csp3) and flexibility (No of rotatable bonds). Consequently, absorption parameters such as Human Intestinal Absorption, Blood Brain Barrier, P-glycoprotein interaction (substrate or inhibitor) and metabolism (inhibitor or substrate) of the bio-active molecules with different cytochrome P450 enzymes were also estimated using admetSAR, which is based on QSAR data for prediction of absorption, distribution, metabolism, excretion and toxicity (ADMET). Drug-like molecules considering Lipinski rules and good ADMET properties were chosen as ligands in consequent molecular docking procedure.
2.2.1 SwissADME
Swiss ADME is a free web tool developed by the Swiss Institute of Bioinformatics (SIB), it was employed to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules and freely available at www.swissadme.ch. It calculates six physicochemical properties: lipophilicity, size, polarity, solubility, flexibility, and saturation. In addition, it promotes the assessment of ADME parameters (absorption, distribution, metabolism and excretion) of drug candidates and molecules and provides information to determine Lipinski’s rule of five for drug likeness of oral bioavailability (Daina et al., 2017). The drug-likeness assessment is based on the following factors: molar mass smaller amount than 500 g mol-1, l Log P is a smaller amount than five, number of acceptors of hydrogen bonds is a smaller amount than 10; accounted for in the molecule function of N or O atoms, and number of donors of hydrogen bonds is a smaller amount than 5, accounted for in the molecule function of NH or OH groups. Hence, the molecules outside this range will be unlikely to become orally bioavailable as a drug (Lipinski et al., 2001).
2.3 Chemistry
2.3.1 General procedure for Synthesis of Schiff base (NHM Compounds):
Schiff’s bases were prepared by reacting of one mole of phenylenediamine and two moles of substituted aromatic aldehydes or ketones in 100 ml ethanol. The solution was refluxed for 7hrs under constant stirring. The condensation reaction was carried out by using glacial acetic acid as a catalyst and monitored by thin layer chromatography (TLC). The mixture was left for cooling and thesolid product (crude) was gathered by filtration and washed four times with ethanol and then dried utilizing vacuum. The gained product was re-dissolved in ethanol for recrystallization and after that dried to give a clean pure product (Scheme 1). The crystalline products obtained were characterized by Fourier-Transform Infrared (FTIR) spectra (Cary 600 FTIR, USA), which was operated inawavenumber range of 4000–400 cm−1, and 1H and 13C nuclear magnetic resonance (NMR) spectroscopies were performed with dimethyl disulfide (DMDS) used as solvent. Tetramethylsilane (TMS) was the standard reference, and the probe temperature was 25°C (Hamil et al., 2009).
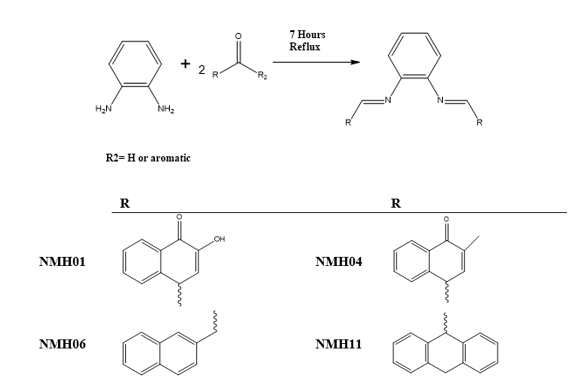
Scheme 1: General scheme for the synthesis of Schiff base NHM Compounds.
((4Z,4'E)-4,4'-(1, 2-phenylenebis (azanylylidene)bis (2-hydroxynaphthalene-1(4H)-one) (NHM01)
NMH01 has been synthesized according to the general procedure, by reacting of one mole of phenylenediamine and two moles of 2-hydroxynaphthalene-1,4-dione in 100 ml ethanol.
Spectral data
Orange crystals, mp 240-241ºC, yield: 62%; %; FTIR (cm-1):3570 (O-H), 1619(C=O) 1586(C=N), 1445(C-N),1216(C-O). 1H NMR (400 MHz, DMSO-d6, δ ppm) 7.84-8.29(m, 12H, C-H aromatic), 7.19 (S, 1H, C-H aromatic), 15(S, 2H, OH). 13CNMR (400 MHz, DMSO-d6):189.10, 157.04, 145.42, 142.65, 139.36, 139.62, 131.07, 130.29,130.16, 129.38, 128.84, 128.60, 128.23, 124.90, 123.00,103.00 .Anal Calcd for C26H16N2O4: C, 74.28; H, /3.84; N, 6.66; O, 15.22.
(4Z,4'Z)-4,4'-(1,2-phenylenebis (azanylylidene)) bis(2-methylnaphthalene-1(4H)-one)NHM04compound:
NMH04 has been synthesized according to the general procedure, by reacting of one mole of phenylenediamine and two moles of 2-methylnaphthalene-1,4-dione in 100 ml ethanol.
Spectral data
Green crystals, mp 241-242ºC, yield: 75%; FTIR (cm-1):3053 (C-H, Aromatic), 2965 C-H (CH3)3570 (O-H), 1600(C=O) 1550(C=N), 1526 (C=C), 1440(C-N),1220(C-O). 1H NMR (400 MHz, DMSO-d6, δ ppm) 7.30-8.05 (m, 12H, C-H aromatic), 6.9 (s, 1H, CH=C aromatic), 2.09 (s, 6H, CH3). 13CNMR (400 MHz, DMSO-d6); 184.81, 147.82,135.02,133.77, 133.74, 131.59, 131.56, 125.82, 125.13, 16.50. Anal Calcd for C28H20N2O2: C, 80.75; H, 4.84; N, 6.73; O, 7.68.
(N1E,N2E)-N1,N2-bis (naphthalen-1ylmethylene)benzene-1,2-diamine) NHM06 compound:
NMH06has been synthesized according to the general procedure, by reacting of one mole of phenylenediamine and two moles of 1-naphthaldehyde in 100 ml ethanol.
Spectral data
White crystals, mp 239-240ºC, yield: 65%; FTIR (cm-1): 3045(C-H aromatic), 1529(C=N),1446(C=C), 1397(C-N) 1H NMR (400 MHz, DMSO-d6, δ ppm):7.32-8.05 (m, 18H,C-H aromatics),9.1(s, 2H, CH=N). 13CNMR (400 MHz, DMSO-d6); 156.00,140.41, 135.18,134.84,133.42, 133.36,130.42, 130.31, 127.88,127.68,126.63,124.13,116.39. Anal Calcd for C28H20N2: C, 87.47; H, 5.24; N, 7.29.
(N1,N2-di(anthracen-9(10H)-ylidene)benzene-1,2-diamine) NMH11 compound:
NMH11 has been synthesized by the described procedure, by reacting of one mole of phenylenediamine and two moles of anthracen-9(10H)-one in 100 ml ethanol.
Spectral data
Light green crystals, mp 291-292ºC, yield: 80%; FTIR (cm-1):3287(C-H aromatic), 3060(C-H SP3), 1655 (C=N), 1577(C=C), 1308(C-N). 1H NMR (400 MHz, DMSO-d6, δ ppm) 3.81(s, 4H,CH2), 7.30-7.72 (m, 20H, C-H aromatics). 13CNMR (400 MHz, DMSO-d6); 185.01,148.62, 140.85, 139.86, 134.35, 132,81, 131.94, 128.91, 127.68, 127.59, 126.55, 125.37, 125.34, 123.44, 122.35 119.74, 116.43, 50.01. Anal Calcd for C34H24N2: C, 88.67; H, 5.25; N, 6.08.
2.4 DNA binding studies
To study how competently the synthesized compounds interact with G-DNA, the synthesized compounds were investigated for their DNA binding ability using fluorescence emission spectra. All experiments were conducted in Tris (Hydroxymethyl)aminomethane (Tris buffer) (0.01M Tris, 0.1M NaCl, at pH 7.4), glass-distilled deionized water and analytical grade reagents were used throughout experiments. The pH values of all solutions were measured with a calibrated Jenway pH-meter model 3510 (Staffordshire, UK). All buffer solutions were filtered through Millipore filters (Millipore, UK) of 0.45 mm pore diameter.
2.4.1 Absorbance spectra
Absorbance spectra were measured on a Jenway UV-visible spectrophotometer, model 6505 (London, UK) using quartz cells of 1.00cm path length. The UV-Vis absorbance spectra were recorded in the 200-500nm range, and spectral bandwidth of 3.0nm. For the final spectrum of each solution analyzed baseline subtraction of the buffer solution was performed. Genomic DNA was used in a concentration of 75 μg/ml. DNA was extracted from peripheral lymphocytes of anticoagulated blood (EDTA) samples by Proteinase K digestion and phenol/chloroform extraction.Purity was determined by measuring the absorbance at 260/280nm indicating that the sample is free from protein contamination. The concentration was assayed spectrophotometrically using 6600M-1cm-1 as a molar extinction coefficient at 260nm.
2.4.2 Fluorescence spectra and DNA-binding studies
Fluorescence emission and excitation spectra were measured using a Jasco FP-6200 spectrofluorometer (Tokyo, Japan) using fluorescence 4-sided quartz cuvettes of 1.00 cm path length. The automatic shutter-on function was used to minimize photo bleaching of the sample. The selected excitation wavelength for ethidium bromide was 480nm. The emission spectrum was corrected for background fluorescence of the buffer. The ethidium bromide (EB) fluorescence displacement experiment was performed by sequential addition of aliquots of 1790 μlTris buffer, 10 μl EB (final concentration of 72μM), 100μl G-DNA from stock solutions (1.5 mg/ml) and finally 10 μl of compounds (NHM compounds, final concentration of 30 μM). Emission spectra were recorded for each system using excitation wavelengths of maximum fluorescence intensity determined for the systems to be 480nm using a slit width of 5nm to examine alterations in emission spectra resulting from the complex construction of both systems. On construction of the full systems, the system was allowed to equilibrate for 30minutes at room a temperature and emission spectra (500-730nm) were recorded to monitor changes in EB intensity.
3.1 Design strategy of ortho-phenylenediamineSchiff’s base derivatives
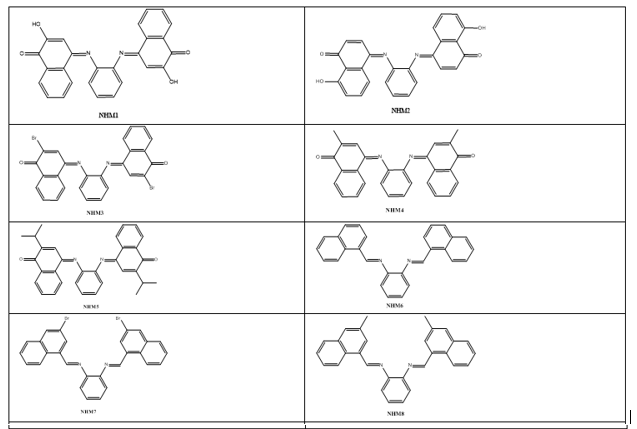
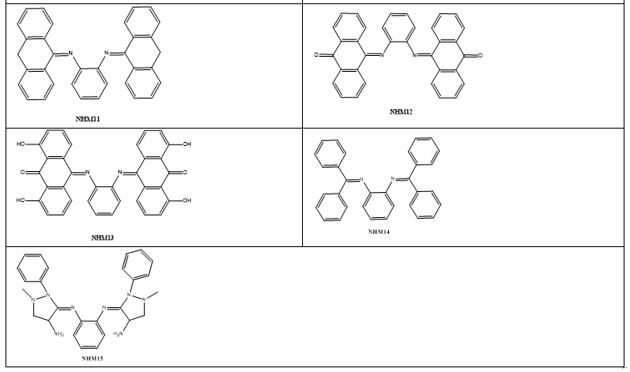
Based on the increased role and importance of phenylenediamineSchiff’s base derivatives as DNA intercalators as described in the introduction part, and because of their versatile route of chemical synthesis, ortho-phenylenediamine were used as key scaffold to develop a series of aromatic imino analogues. Fifteen compounds named with NHM symbol were designed, the strategy which applied to design these compounds was based on the symmetrical distribution of the amino groups of ortho-phenylenediamine with different carbonyl compounds including naphthaldehyde, naphathquinone and anthraquinone derivatives, where the aldehydes or ketones differ in their lipophilicity, hydrogen bond ability as donor or acceptors, and ionizability. The applied strategy yielded the scaffold which contains three parts: 1) A planar polycyclic pattern to intercalate between DNA base-pairs and to provide π-π interactions. 2) Some polar functions providing hydrogen bond interactions with nucleic acids. 3) Variety of substituents to improve the interactions and promote the physicochemical properties. To the best of our knowledge, there have been no literature reports regarding Schiff’s base of ortho-phenylenediamine with naphthoquinones and anthraquinone derivatives as DNA intercalating agent so far. The designed compounds were investigated for their docking energies. In addition, the effect of replacing of the naphthyl ring with a three ring system (anthracene) or aromatic rings with flexible linkage was also investigated. The chemical structures are listed in Table 1.
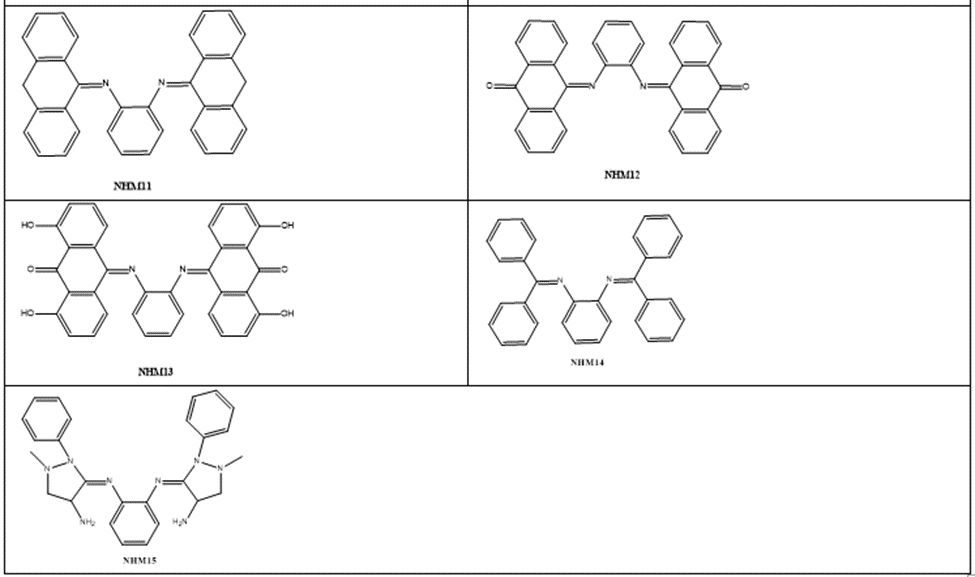
Table 1: The chemical structures of designed compound
3.2. Molecular docking analysis
In this part of the current study, the molecular dockings of NHM compounds with five B-DNA fragments were performed using Auto Dock vina in PyRx Virtual Screening Tool to understand the possible binding mode. The molecular docking study was employed to identify the binding interactions and estimate the binding affinity of the most active derivatives to the DNA duplexby combining and optimizing variables such as hydrophobic, steric, and electrostatic complementation. Two well-known biologically active DNA intercalating agents were used as references namely doxorubicin and daunorubicin. Considering the values of free energies of binding mentioned in Table 2, it could be deduced that the compounds were properly accommodated between the DNA base pairs. The molecular docking analysis revealed that the compounds have contributed in hydrophobic and Pi-Pi interactions, and hydrogen bonding with the DNA base pairs.
| Names of compound | Docking Energy(kcal/mol) | ||||
| 1D60 | 1ZEW | 2DES | 1BNA | 1DC0 | |
| Daunorubicin | -8.9 | -10.0 | -8.8 | -8.9 | -9.8 |
| Doxorubicin | -8.6 | -9.8 | -8.4 | -8.8 | -9.6 |
| NHM01 | -9.2 | -10.9 | -9.1 | -8.9 | -8.8 |
| NHM02 | -7.7 | -9.4 | -9.1 | -8.6 | -8.8 |
| NHM03 | -6.8 | -8.5 | -8.4 | -7.3 | -8.1 |
| NHM04 | -7.3 | -8.1 | -8.7 | -6.9 | -8.0 |
| NHM05 | -6.9 | -8.2 | -9.7 | -6.5 | -8.0 |
| NHM06 | -7.3 | -8.1 | -7.7 | -8.1 | -7.2 |
| NHM07 | -7.3 | -8,3 | -8.0 | -7.3 | -7.2 |
| NHM08 | -7.0 | -8.6 | -9.0 | -7.5 | -7,3 |
| NHM09 | -8.6 | -8.3 | -8.6 | -8.1 | -7.3 |
| NMH10 | -7.2 | -7.8 | -7.6 | -7.7 | -7.6 |
| NMH11 | -8.9 | -9.1 | -8.4 | -8.4 | -8.5 |
| NMH12 | -8.3 | -9.4 | -9.3 | -8.5 | -8.8 |
| NMH13 | -8.9 | -9.0 | -9.4 | -8.5 | -8.8 |
| NMH14 | -6.5 | -7.2 | -6.4 | -6.3 | -7.1 |
| NMH15 | -6.0 | -7.2 | -6.3 | -6.7 | -7.4 |
Table 2: Various energies in the binding process of NHM compounds with DNAs obtained from molecular docking.
(1BNA): D(CGCGAATTCGCG)2, (1DC0): D(CATGGGCCCATG)2, ((2DES): D(CGTACG)2, (1D60) :D(CCAACNTTGG)2, (1ZEW): D(CCTCTAGAGG)2,
ΔGa is the binding free energy change in the binding process.
The more negative the numerical values for the binding affinity, the better is the predicted binding between a ligand and a macromolecule. In this particular case of screening five DNA fragments (1BNA, 1ZEW,2DES,1D60, 1BNA and 1DCO) with designed compounds. Table 2 shows that all the designed compounds have good binding energies when compared to standard drugs. Moreover, NMH01 and NMH11 are both predicted to have the best binding affinity.
The DNA fragment (1zew) was used to discuss and compare the intercalating activity of all compounds. It appears worth to note that compounds which exhibit high activity against (1zew) also showed high activity against the other DNA fragments. Simultaneously, any modification which might decrease the activity against (1zew) will produce the same effect against all other DNA fragments.
Molecular docking results were identified based on the strongest ligand binding poses, where identified using the low binding energy and the number of H-bonding and hydrophobic interactions. From the docking studies the molecular docking analysis revealed that all the docked molecules were inserted between the adjacent nucleotides at the active site. These compounds were stabilized at the active site through different interactions. The planar system was involved in hydrophobic stacking with the base pairs of different nucleotides, and the hydrophilic moieties formed several hydrogen bonds with different nucleotides.
It was observed that introduction of hydrophilic substituent's to the aromatic ring increases the intercalating activity (decrease of the docking energy) as shown with NMH01,NMH09 and NMH13. While, the introduction of hydrophobic substituents (R) to the same aromatic ring leads to a decrease of the intercalating activity (increase ofthe docking energy) as shown for NMH04 and NMH08. A substantial observation was that the highest activity was predicted to be with an introduction of 2-hydroxynaphthalene-1,4-dione at NMH01. The importance of hydroxyl substituents at meta-positions of the aromatic ring are attributed to the formation of seven hydrogen bonds at intercalating sites keeping the planarity which is needed for optimum localization at the intercalating site (Figure1). Whereas, hydroxyl substituent at position five builds three H-bonds as shown in NMH02.H-bonding characteristics of compounds rendered significantly high binding affinity with DNA.
Lipophilic substituents (Cl or Br) at position 3 of the naphthyl side chain had little effect on the intercalating activity when compared with the non-substituted derivative (NMH03, NMH07). Furthermore, introduction of a hydrogen bond acceptor substituent such as carbonyl group on the aromatic ring leads to an increase of the intercalating activity, as could be seen from the comparison of 2-hydroxynaphthalene-1,4-dione derivatives with the corresponding non-substituted 2-hydroxynaphthalene derivatives as shown with NMH01,NMH02 and NMH09. In the case of compounds NMH04, NMH08,NMH05 and NMH10 replacement of the methyl moiety on aromatic rings with the isopropyl moiety leads to a decrease of the intercalating activity, which may be caused by steric hindrance of branched groups. Furthermore, as observed in the docking study, compound NMH14 was located at the groove pocket interacting with DNA by pi-anion interaction with DGB:17and DGA:7. Compound NMH15 was located at the groove pocket and interacted with DNA by six hydrogen bonds with DGA:10, DGA:9 DTB:15 and DAB:16. Nevertheless, it was evident that NMH14 and NMH15 had the lowest binding energy scores which may be due to the lack of the planarity pattern of the polyaromatic systems.
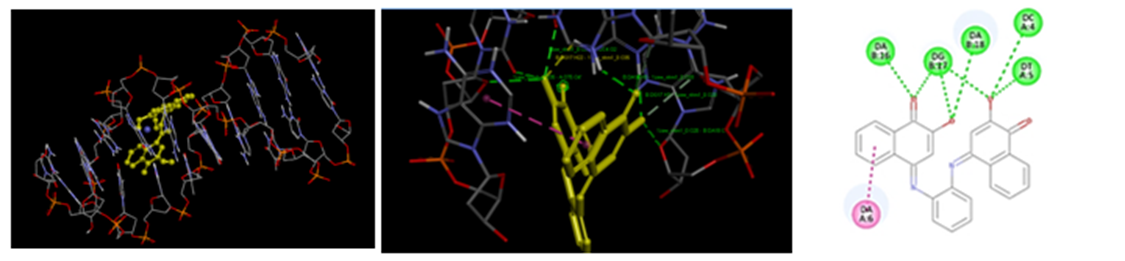
Figure 1: Localization of NHM01 (yellow) at the intercalating site of DNA. (ii)Compound NMH01 interacting with DNA strands with seven intermolecular hydrogen bonds. (iii) 2-dimensional structure of NMH01 at the active site.
It can be clearly seen that the interaction mode of most compounds consist of a classical intercalation mode except the binding of NMH03, NMH05, NMH08 and NMH10 with DNA which involves partial intercalation and groove binding. Moreover, compounds NMH14 and NMH15 bind to DNA via groove binding. It is worth mentioning that compounds NMH01, NMH04, NMH06 and NMH11 were the most active compounds for each DNA fragment (Table 2). For this reason they have been selected for further synthesis and analysis.
3.3 ADMET properties
3.3.1 Analysis of Physicochemical Properties.
General characteristics of designed Schiff’s base derivatives of o-phenylenediamine revealed that all these compounds have a molecular weight less than 500 g/mol, except NMH03, NMH07, and NMH13 with molecular weights of 546.21, 542.26 and 552.53g/mol, respectively. This appears a prime property to be called as drug likeness of the small molecules. It is evident from Table 3 topological polar surface area (TPSA) values of all compounds were found to be within the limit of ≤ 140 Å. The prediction of TPSA parameters helps to understand the passive molecular transport of drug molecules. TPSA is an important tool in drug discovery and development. By analyzing a drug candidate's TPSA, we can predict its potential for oral bioavailability and ability to reach target sites within the body. This prediction hinges on a drug's ability to permeate biological barriers. The Molar refractivity (MR) of all compounds is within the ranges of 121.50–158.01, their number of rotatable bond (nRotb), number of H-bond donors (nHBD) and the number of hydrogen bond acceptors (NHBAs) of all o-phenylenediamine derivatives are within the recognized limits ≤10, ≤5, and ≤10, respectively.
SwissADME predictions suggest that symmetrical phenylenediamine Schiff’s base derivatives have optimum parameters for anticancer activity and can be considered as lead molecules for further modifications.
| Compound | MW (g/mol) | nHA | nRB | nHBD | nHBA | MR | TPSA (A2) |
| NHM01 | 420.42 g/mol | 32 | 2 | 2 | 6 | 121.80 | 99.32Ų |
| NHM02 | 420.42 g/mol | 32 | 2 | 2 | 6 | 122.71 | 99.32Ų |
| NHM03 | 546.21 g/mol | 32 | 2 | 0 | 4 | 134.40 | 58.86Ų |
| NHM04 | 416.47 g/mol | 32 | 2 | 0 | 4 | 128.27 | 58.86Ų |
| NHM05 | 472.58 g/mol | 36 | 4 | 0 | 4 | 147.50 | 58.86 Ų |
| NHM06 | 384.47 g/mol | 30 | 4 | 0 | 2 | 128.85 | 24.72Ų |
| NHM07 | 542.26 g/mol | 32 | 4 | 0 | 2 | 144.25 | 24.72Ų |
| NHM08 | 412.52 g/mol | 32 | 4 | 0 | 2 | 138.78 | 24.72Ų |
| NHM09 | 416.47 g/mol | 26 | 4 | 2 | 4 | 132.89 | 65.18Ų |
| NMH10 | 468.63 g/mol | 36 | 6 | 0 | 2 | 158.01 | 24.72Ų |
| NMH11 | 460.57 g/mol | 36 | 2 | 0 | 2 | 148.83 | 24.72Ų |
| NMH12 | 488.53 g/mol | 38 | 2 | 0 | 4 | 149.67 | 58.86Ų |
| NMH13 | 552.53 g/mol | 42 | 2 | 4 | 8 | 157.76 | 139.78Ų |
| NMH14 | 436.55 g/mol | 34 | 6 | 0 | 2 | 142.81 | 24.72Ų |
| NMH15 | 454.57 g/mol | 34 | 4 | 2 | 6 | 152.22 | 89.72Ų |
Table 3: Physicochemical properties of designed Schiff’s base derivatives of o-phenylenediamine calculated using swissADME database.
Molecular weight: M. W, Number of heavy atom: nHA, , Number of rotatable bonds: nRB, Number of H-bond acceptors: nHBA, Number of H-bond donors: nHBD, Molar refractivity: MR, topological polar surface area: TPSA.
3.3.2 Lipophilicity and Water Solubility.
Log Po/w values of all derivatives range from 2.85 to 8.00 reflecting its partition preferably into the lipid compartment (Table 4). This implies that
these compounds will be well absorbed through the membranes into the systemic circulation to achieve high bioavailability. On the other hand, solubility, Log Sis aqueous solubility, exhibit a defined range of -4.4to-8.72 as shown in Table 5. The compounds NMH01, NMH02, NMH03, NMH15 predicted as moderately soluble (with optimal lipophilicity and moderate water solubility) can achieve good bioavailability when administered orally. All other derivatives are poorly water soluble.
| Compound | iLOGP | XLOGP3 | WLOGP | MLOGP | SILICOS-IT | Consensus Log Po/w |
| NHM01 | 2.62 | 4.36 | 5.20 | 1.25 | 5.06 | 3.70 |
| NHM02 | 3.15 | 5.04 | 4.84 | 1.79 | 5.10 | 3.98 |
| NHM03 | 3.20 | 6.45 | 6.88 | 3.49 | 7.37 | 5.48 |
| NHM05 | 4.13 | 6.92 | 7.49 | 4.06 | 8.35 | 6.19 |
| NHM06 | 4.13 | 7.02 | 7.49 | 5.35 | 7.89 | 6.38 |
| NHM07 | 4.09 | 8.40 | 9.02 | 6.46 | 9.22 | 7.44 |
| NHM08 | 4.04 | 7.75 | 8.11 | 5.74 | 8.94 | 6.92 |
| NHM09 | 3.57 | 6.31 | 6.91 | 4.15 | 6.91 | 5.57 |
| NMH10 | 4.32 | 9.27 | 9.74 | 6.48 | 10.21 | 8.20 |
| NMH11 | 4.26 | 8.46 | 7.83 | 6.03 | 9.19 | 7.15 |
| NMH12 | 3.62 | 7.49 | 7.11 | 4.17 | 8.56 | 6.19 |
| NMH13 | 3.88 | 7.17 | 5.91 | 2.01 | 6.63 | 5.13 |
| NMH14 | 3.89 | 8.28 | 8.02 | 6.05 | 8.40 | 6.93 |
| NMH15 | 3.62 | 2.58 | 1.61 | 3.73 | 1.08 | 2.52 |
Table 4: The lipophilicity of designed Schiff’s base derivatives of o-phenylenediamine calculated with SwissADME database.
| Compound | ESOL | Ali | SILICOS- IT | |||
Log S (ESOL) | Class | Log S (Ali) | Class | Log S SILICOS-IT | Class | |
| NHM01 | -5.48 | Moderately soluble | -6.16 | Poorly soluble | -7.75 | Poorly soluble |
| NHM02 | -5.91 | Moderately soluble | -6.87 | Poorly soluble | -7.31 | Poorly soluble |
| NHM03 | -7.57 | Poorly soluble | -7.48 | Poorly soluble | -10.47 | Insoluble |
| NHM04 | -6.11 | Poorly soluble | -6.39 | Poorly soluble | -9.68 | Poorly soluble |
| NHM05 | -7.24 | Poorly soluble | -7.97 | Poorly soluble | -10.49 | Inoluble |
| NHM06 | -7.02 | Poorly soluble | -7.35 | Poorly soluble | -10.90 | Insoluble |
| NHM07 | -8.83 | Poorly soluble | -8.79 | Poorly soluble | -12.44 | Insoluble |
| NHM08 | -7.62 | Poorly soluble | -8.11 | Poorly soluble | -11.65 | Insoluble |
| NHM09 | -6.73 | Poorly soluble | -7.47 | Poorly soluble | -9.73 | Poorly soluble |
| NMH10 | -8.72 | Poorly soluble | -9.69 | Poorly soluble | -12.47 | Insoluble |
| NMH11 | -8.51 | Poorly soluble | -8.85 | Poorly soluble | -13.50 | Insoluble |
| NMH12 | -8.04 | Poorly soluble | -8.56 | Poorly soluble | -13.24 | Insoluble |
| NMH13 | -8.18 | Poorly soluble | -9.93 | Poorly soluble | -10.87 | Insoluble |
| NMH14 | -8.02 | Poorly soluble | -8.66 | Poorly soluble | -12.54 | Insoluble |
| NMH15 | -4.41 | Moderately soluble | -4.41 | Moderately soluble | -5.97 | Moderately soluble |
Table 5: Water solubility characteristics of designed Schiff’s base derivatives of o-phenylenediamine calculated with swissADME database
3.3.3 Pharmacokinetic Profile
Values of ADME properties of designed o-phenylenediamine derivatives presented in Table 6 were all within the permissible range. All of the compounds have a low gastro intestinal absorption except NMH01, NMH02, NMH04 and NMH15 show high gastrointestinal absorption. All
these compounds lack blood brain barrier (BBB) penetration. BBB is very important to limit the penetration of drugs into the central nervous system (CNS). The lack of BBB penetration ability suggests their low CNS side effects as well as their unsuitability to treat brain tumors'. In the present study, all the compounds were predicted to be non-substrate of P-glycoprotein, except compounds NMH06, NMH07, NMH08, NMH09, NMH10, NMH14 and NMH15. In the case of drug metabolism NMH01 was identified as an inhibitor of CYP1A2, CYP2C9 and CYP3A4. Compound NMH04 was predicted as an inhibitor of CYP2C9, CYP2C19 and CYP3A4 and in addition, NMH06 as CYP1A2, CYP2C19 and CYP3A4 inhibitors. However, NMH11 was predicted to show no inhibitory activity against any of the CYP isoforms. The skin permeability coefficient (Log Kp, a multiple linear regression, Potts and Guy, 1992), indicates the more negative the log Kp (withKpin cm/s), the less skin permeant is the molecule. Among the NHM compounds, NMH15 (-7.24 cm/s) is the least permeant compound and NMH10 (-2.58 cm/s) is most permeant, respectively.
| Compound | Absorption | BBB Permeant | P-gp substrate | CYP1A2 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor | Log Kp (Skin Permeation) (cm/s) |
| NHM01 | High | No | No | Yes | No | Yes | No | Yes | -5.77 cm/s |
| NHM02 | High | No | No | Yes | Yes | Yes | No | Yes | -5.29 cm/s |
| NHM03 | Low | No | No | No | No | Yes | No | No | -5.05 cm/s |
| NHM04 | High | No | No | No | Yes | Yes | No | Yes | -5.01 cm/s |
| NHM05 | Low | No | No | No | No | No | No | Yes | -4.27 cm/s |
| NHM06 | Low | No | Yes | Yes | Yes | No | No | Yes | -3.66 cm/s |
| NHM07 | Low | No | Yes | Yes | No | No | No | No | -3.64 cm/s |
| NHM08 | Low | No | Yes | Yes | No | No | No | Yes | -3.31 cm/s |
| NHM09 | Low | No | Yes | Yes | Yes | No | No | No | -4.36 cm/s |
| NMH10 | Low | No | Yes | No | No | No | Yes | Yes | -2.58 cm/s |
| NMH11 | Low | No | No | No | No | No | No | No | -3.10 cm/s |
| NMH12 | Low | No | No | Yes | No | No | No | No | -3.96 cm/s |
| NMH13 | Low | No | No | No | No | No | No | No | -4.58 cm/s |
| NMH14 | Low | No | Yes | Yes | Yes | No | No | Yes | -3.08 cm/s |
| NMH15 | High | No | Yes | No | No | No | Yes | No | -7.24 cm/s |
Table 6: Pharmacokinetic parameters of designed Schiff’s base derivatives of o-phenylenediamine calculated with swissADME database.
3.3.4 Drug likeness
NMH01 do not violate any of the five drug likeness parameters, i.e., Lipinski, Muegge, Ghose, Veber, and Eganrules of drug likeness(Daina et al., 2017). The other compounds showed violation of at least one of the drug-likeness filters. As per Lipinski's rule-of-five, candidates violating none or less than one of the following four criteria are likely to be developed as a prospective oral drug. The criteria are as follows: log P (octanol-water partition coefficient) ≤5, molecular weight ≤500, number of hydrogen bond acceptors ≤10 and number of hydrogen bond donors ≤5. In this investigation, none of the designed Schiff’s base derivatives of o-phenylenediamine compounds has violated Lipinski's rule of five except NMH07 which has two violations: MW>500 and MLOGP>4.15 (Table 7) thus providing the possible utility of as eries for developing compounds with drug-like properties. All derivatives have a similar bioavailability score of 0.55. The results obtained from in silico studies using SwissADME clearly indicate that the selected compounds to be synthesized, namely NMH01, NMH04, NMH06 and NMH11 could be druggable substances. It is interesting to note that the results from the SwissADME predictor values of MW1, log P, HBA, HBD, molar refractivity and the total polar surface area of these molecules are in excellent agreement with the most important rules of drug likeness. The results show that all four compounds have zero violations of Lipinski’s rule of five. SwissADME predictions suggest that symmetrical phenylenediamine Schiff’s base derivatives have optimum parameters for anticancer activity and can be considered as lead molecules for further modifications.
| Compound | Lipinski | Hose | Veber | Egan | Muegge | BA. Score |
| NHM01 | Yes | Yes | Yes | Yes | Yes | 0.55 |
| NHM02 | Yes | Yes | Yes | Yes | No;1violation: XLOGP3>5 | 0.55 |
| NHM03 | Yes;1violation: MW>500 | No; 3 violations: MW>480,WLOGP>5.6, MR>130 | Yes | No;1violation: WLOGP>5.88 | No;1violation: XLOGP3>5 | 0.55 |
| NHM04 | Yes | No;1violation: WLOGP>5.6 | Yes | No;1violation: WLOGP>5.88 | No;1violation: XLOGP3>5 | 0.55 |
| NHM05 | Yes | No; 2 violations: WLOGP>5.6,MR>130 | Yes | No; 1 violation: WLOGP>5.88 | No;1violation: XLOGP3>5 | 0.55 |
| NHM06 | Yes;1violation: MLOGP>4.15 | Yes | No;1violation: WLOGP>5.88 | No;1violation: WLOGP>5.88 | No;1violation: XLOGP3>5 | 0.55 |
| NHM07 | No;2violations: MW>500,MLOGP>4.15 | No;3violations: MW>480,WLOGP>5.6, MR>130 | Yes | No;1violation: WLOGP>5.88 | No;1violation: XLOGP3>5 | 0.17 |
| NHM08 | Yes; 1 violation: MLOGP>4.15
| No; 2 violations: WLOGP>5.6,MR>130 | Yes | No;1violation: WLOGP>5.88 | No;1violation: XLOGP3>5 | 0.55 |
| NHM09 | Yes;1violation: MLOGP>4.15 | No;2violations: WLOGP>5.6,MR>130 | Yes | No;1violation: WLOGP>5.88 | No;1violation: XLOGP3>5 | 0.55 |
| NMH10 | Yes;1violation: MLOGP>4.15 | No;2violations: WLOGP>5.6,MR>130 | Yes | No;1violation: WLOGP>5.88 | No;1violation: XLOGP3>5 | 0.55 |
| NMH11 | Yes;1violation: MLOGP>4.15 | No;2 violations: WLOGP>5.6,MR>130 | Yes | No;1 violation: WLOGP>5.88 | No;1 violation: XLOGP3>5 | 0.55 |
| NMH12 | Yes;1violation: MLOGP>4.15 | No; 3 violations: MW>480,WLOGP>5.6, MR>130 | Yes | No; 1 violation: WLOGP>5.88 | No;1 violation: XLOGP3>5 | 0.55 |
| NMH13 | Yes;1violation: MW>500 | No;3violations: MW>480,WLOGP>5.6, MR>130 | Yes | No; 2 violations: WLOGP>5.88,TPSA>131.6 | No;1 violation: XLOGP3>5 | 0.55
|
Table 7: Druglikenessof designed Schiff’s base derivatives of o-phenylenediamine calculated with swissADME database
3.4 Chemical synthesis
The best four promising compounds according to the in silico studies were synthesized. The targeted compounds, symmetrical 1, 2-phenylenediamine with di-substituted aromatic carbonyl moiety, have been successfully synthesized using a previously reported synthetic procedure. The reaction order for the synthesis of selected compounds is given in general schemes 2. All the derivatives were synthesized producing good yields with characteristic color. The compounds were soluble in chloroform and dimethyl sulfoxide (DMSO) but insoluble in water. Melting points were in the range of 239–292 °C. Thin-layer chromatography (TLC) was used to monitor the progress of the reaction in methanol: dichloromethane (1:4). Their chemical structures were confirmed by means of spectroscopic analysis as described previously.
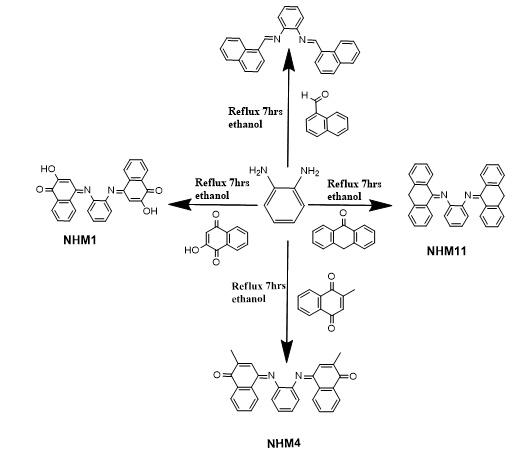
Scheme 2: Synthesis scheme of NHM compounds
Analysis of spectra
Proton 1H andCarbon 13CNMR spectra
NMR analysis is very important to confirm the structure and functional groups. Experimental H-NMR chemical shifts were expressed in ppm, dispersed through the spectrum starting form tetramethylsilane as the internal standard and recorded in DMSO-d. Importantly, the spectra lacked the signal of the amino group (NH) characteristic of the starting material. The ¹H-NMR spectrum of NHM01 displayed singlet at 15 ppm and multiplet at 7.84-8.29 ppm as signal to the O-H proton and the protons of aromatic rings, respectively. Moreover, a peak appears at 2.09 ppm attributed to the aliphatic–CH (6H) compound contained in NHM04 and multiplet signals at 7.30-8.05 ppm were ascribed to aromatic protons. Besides, the signal due to –CH=N appears at 9.1 ppm for the compound NHM06 corresponding to the azomethine group which confirms the formation of Schiff bases and the multiple peaks attributed to the aromatic protons between 7.32 and 8.05 ppm. Additionally, the ¹H-NMR spectrum of compound NMH11 exhibited as singlet signal at 3.81 ppm corresponding to the CH2 group. The multiplet signals at 7.30-7.72 ppm were assigned to the 20 H aromatic protons. Consistently13C-NMR spectra showed a chemical shift at 189.10, 184.81,156.40,185.01 ppm assigned to the imine group revealing the formation of Schiff's base derivatives of o-phenylenediamine (NHM01, NHM04, NHM06 and NMH11), respectively. In addition, the peak observed at 16.50 ppm was attributed to a methyl carbon (C20) integrated at the aromatic group of compound NHM04. The peaks observed at 157.04 ppm and 183.00 ppm corresponding to carbonyl groups of the compounds NMH01 and NMH04, respectively.The peak at 50.01ppm attributes to the methylene group of compound NMH11. The peaks at 103.11-148.62 ppm range are ascribed to aromatic carbons.
Fourier-Transformed Infrared
In order to continue the characterization of NHM compounds, the FT-IR spectrum was carried out. The FT-IR spectra of NHM compounds showed the disappearance of both carbonyl groups atthe aldehyde, keton or amino groups atthe amine, whereas NH is vanished or hidden underneath the broad bands at3450-3300cm-1 in Schiff’s base. Additionally, the presence of the azomethine CH=N stretching at about 1529−1655 cm-1 depending on the nature of R side chain substituents, indicates the formation of the Schiff base compounds. The FTIR spectrum of the compound NMH01 showed a stretched band around 3200 − 3570 (O-H) cm-1, which is attributed to the intramolecular hydrogen bond of the hydroxyl group. Furthermore, compound NHM01 exhibits absorption bands at 1675-1619cm-1 corresponding to the C=C and C=O, respectively. Additionally, the band at 1446−1450-1577 cm-1 corresponds to C=C of compounds NHM06, NHM04 and NMH11, respectively.
DNA binding studies
UV Absorbance Spectra
UV-spectroscopy being an important qualitative and quantitative analysis technique has been exploited to investigate the interaction of ligands with DNA. The electronic absorption spectrum of DNA exhibits a broad band in the range of 200-350nm and shows maximum absorption at ~260nm. This maximum is a consequence of chromophoric groups in purine and pyrimidine moieties responsible for the electronic transitions (González-ruiz et al., 2011). Therefore, the changes in the absorption spectra of the compound alone and compound–DNA mixture is an indication of DNA–compound interaction, and the mode and strength of binding can also be determined from the shift. The absorbance spectra of DNA could show hypochromism (deceased absorbance intensity) and hyperchromism (increased absorbance intensity) upon titration with the ligand. In general, the intercalative mode of binding usually results in hypochromism and a red-shift because of the strong stacking interaction between an aromatic chromophore and the nitrogen bases of DNA. This results in structural changes of the DNA that entailed the local unwinding of the double helix. Whereas for weak interaction such as hydrogen bonding, groove binding and electrostatic interaction no significant shift of the absorption maxima occurs (Banerjee et al., 2016). The absorption spectra resulting from titrations are depicted in Fig. 2. As observed from spectral curves, the complexes showed pronounced hypochromism with a slight red shift of 2nm. The results suggest that these compounds bind to DNA through an intercalation mode.
The resultant hypochromic effect was found to be 0.244nm for NMH01, 0.201nm for NMH04, 1.53nm for NMH06, and 1.42nm for NMH11 followed by a red shift of magnitude 2.01nm, 1.89nm, 1.73nm and 1.88nm for NMH01, NMH04, NMH06, and NMH11, respectively.
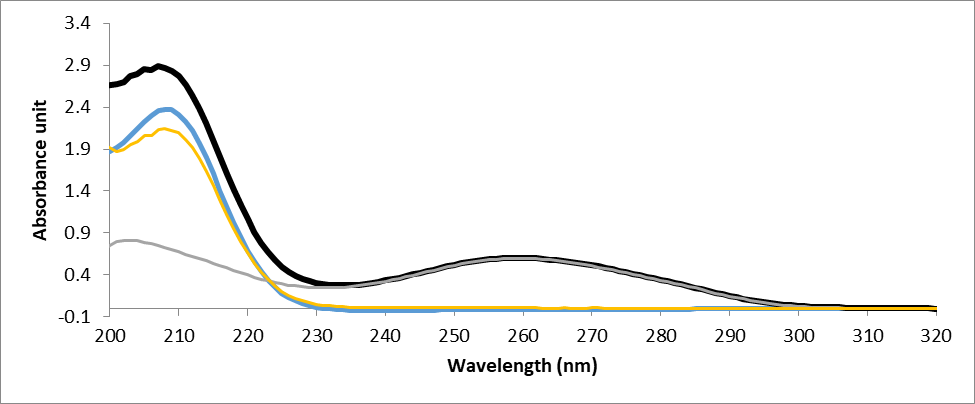
Figure 2: UV–Vis absorption spectra of DNA in the absence and presence of NMH01 (A) the G-DNA-NMH01 complex, (B) the 30 µm NMH01, (C) G-DNA-NMH01 complex subtracted from the G-DNA absorbance to show the hypochromic shift (D) GDNA alone. The experiment was conducted in Tris buffer solution (0.01M Tris, 0.1M NaCl, at pH 7.4). Genomic DNA was used in a concentration of 75 μg/ml and NMH01 30 µM.
Ethidium bromide (EB) Competition Assay
A competitive displacement assay was conducted to obtain further evidence of the binding pattern of the tested compounds and DNA. Ethidium bromide (EB) is a strong fluorescent dye, known to bind to DNA through intercalation with maximum emission at 661 nm upon binding to double stranded DNA. Free Ethidium bromide exhibits very low fluorescence. However, with the addition of DNA, due to its intercalation, the fluorescence intensity magnifies remarkably (Zhao et al., 2013).The gradual addition of compounds 1, 4, 6, and 11 to the EB–DNA complex cause a decrease of the emission (quenching effect) (Figure 3). The hypochromic shift appears as a result of formation of a complex between compound and DNA and this reduction of the fluorescence intensity is a characteristic sign of intercalation. The fluorescence spectra of the EB-DNA complex in the absence and presence of NMH01 is shown in Fig. 3. The figure shows that the fluorescence intensity of EB-DNA can be quenched significantly by gradual addition of NMH01. This result indicates that NMH01 was able to replace EB in the DNA helix indicating an intercalative NMH01 DNA binding mode.
The fluorescence quenching data were analyzed by the Stern–Volmer equation and the quenching constants (Ksv) were calculated in which the synthesized compounds were the quenchers:
I0/I = 1 + KSV [Q] (1)
I0 and I represent the fluorescence intensities in the absence and presence of quencher, respectively; KSV is the linear Stern-Volmer quenching constant; Q is the concentration of quencher. The KSV values were given by the ratio of the slope to intercept. The KSV value suggests a strong affinity of the compounds to EB-bound G-DNA and that it can competitively displace EB from DNA via an intercalative mode of binding. As shown is Table 8, compound NMH01 had the highest KSV value suggesting that compound NMH01 bound most strongly to G-DNA even more than daunurobicin. The KSV values for the tested compounds are listed in Table 8.
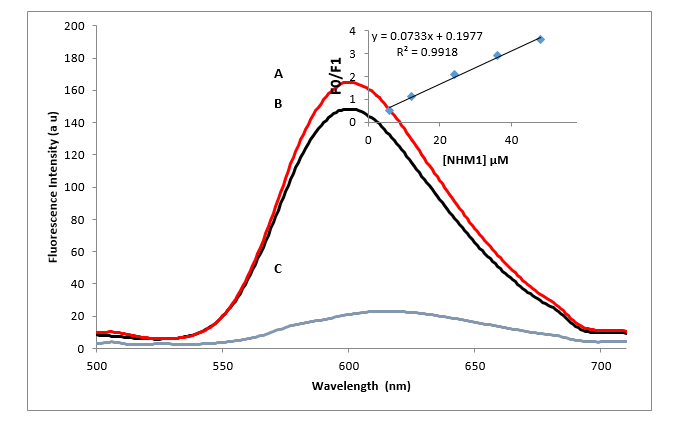
Figure 3. Fluorescence changes of the NMH01 system that contains (A) the G-DNA-ethidium bromide complex, (B) the G-DNA-ethidium bromide complex with 30 µm NMH01 and (C) ethidium bromide alone. The experiment was conducted in Tris buffer solution (0.01M Tris, 0.1M NaCl, at pH 7.4), λex = 480 nm. Inset: plot of F0/F versus different concentrations of NMH01 (µM) for the titration of NMH01 to G-DNA-EB complex. Genomic DNA was used in a concentration of 75 μg/ml and ethidium bromide 72 µM.
Based upon the variation in absorbance and the fluorescence intensities the binding constant for the compounds to determine the strength of the interaction of compounds with DNA at 298 K, was determined from fluorescence titration data using the Benesi–Hildebrand (BH) equation:
1/ΔA =1/ (KA.ΔA0) [DNA] +1/(ΔA0) ······ (2)
Here, the difference in fluorescence was denoted by ΔA, ΔA0 is maximum florescence intensity change and [DNA] is the concentration of DNA. By plotting the reciprocal of the difference in fluorescence intensity against the reciprocal of DNA concentration, the Benesi−Hildebrand plot was constructed. To determine the extent of DNA‐binding affinities of compound towards G‐DNA, the intrinsic binding constant (Kb) values were obtained from the linear regression plots of [1/(ΔA0)] vs. {1/[DNA]} M−1, and the ratio of the slope to intercept calculated. The Kb binding constants of compounds are given in Table 8(Benesi & Hildebrand, 1949).
The Schiff’s base derivatives of o-phenylenediamine compounds showed similar binding constants and these areclose to the binding affinity of the intercalating agent daunorubicin (7.2x104 ±0.004M-1 ), which agrees well with those reported in the literature (7.8 X104 M-1 ) (Hajian et al., 2009). The overall measured Kb findings for all compounds were in the subsequent sequence: 1>4 > 6>11.They were within the range of the intercalation mode (K = 1 × 105 × 1011M-1), where intercalative mode of interactions had been assigned (Wolfe et al., 1987;Zhou et al., 2016).
The free energy (ΔG) was calculated by using equation ΔG RT = − ln K (3), where ΔG is the Gibbs free energy, R is the universal gas constant (taken as 2cal/mol K), T is the absolute temperature (298 K), and K is the binding constant. The observed values of ΔG were in the range of-6.12 to -6.44kcalmol-1(Table 8), which also indicates that the compounds spontaneously intercalate to DNA. Compound (NHM01) exhibited the most excellent binding potency compared to others.
| Name | KSV (M-1) | Ka (M-1) | ΔG kcal mol−1 | R2(5 points) |
| Taxol |
5.90X103 | 5.3 X104 ±0.002 | -6.44 | 0.99 |
| Daunorubicin | 7.91x103 | 7.2 X104±0.004 | -6.62 | 0.97 |
| NMH01 | 9.00X103 | 5.9 X104±0.006 | -6.50 | 0.99 |
| NMH04 | 6.02X103 | 4.3 X104±0.001 | -6.31 | 0.98 |
| NMH06 | 3.02X103 | 3.9 X104±0.004 | -6.25 | 0.978 |
| NMH11 | 2.86X103 | 3.1 X104±0.002 | -6.12 | 0.968 |
Table 8. The key selection vector (KSV) values, the binding constant (Kb)and the free energy (ΔG)of synthesized Schiff’s base derivatives of o-phenylenediamine.
A new series of fifteen compounds (1-15) possessing ortho-phenylenediamine Schiff’s base scaffold were designed. Next, ADMET studies were conducted by using SwissADME software and all produced compounds satisfied the Lipinski criterion. Furthermore, the results of docking studies revealed that the docked compounds displayed strong binding affinity with DNA and have a similar binding mode to that of daunorubicin (DNA intercalating agent used as reference) with binding energies ranging from -6.3 to -10.9kcal/mol. Among all these compounds, four compounds (NMH01, 04, 06, and 11) have been synthesized and characterized by FT-IR, 1H and 13C NMR spectroscopy. The interaction of these compounds with genomic-DNA (G-DNA) has been explored by using absorption spectral and fluorescence displacement studies using ethidium bromide. The results indicate that the compounds bind strongly to G-DNA via an intercalation mechanismNHM01 showed the highest key selection vector (KSV) value, suggesting that compound NHM01binds most tightly to G-DNA. Therefore, NHM01, NHM04, NHM06 and NMH11are proposed as effective cytotoxic agents with potential DNA intercalation and may serve as lead compounds for the discovery of new anticancer agents. Further biological evaluations are necessary to confirm our findings.
We gratefully acknowledge Garabolle Research Center (GRC) for performing FTIR study.
The authors declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest and is compliant with ethical standards.
This article does not contain any studies with human participants performed by any of the authors.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
