
Case Report | DOI: https://doi.org/10.31579/2641-5194/032
*Corresponding Author: Sean Lee, Endovascular Surgery, Vascular Surgery, USA
Citation: Lee S. (2021) Stomach this: a novel type of gastritis. J. Gastroenterology Pancreatology and Hepatobilary Disorders. 5(6) DOI: 10.31579/2641-5194/032
Copyright: © 2021, Sean Lee, This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 20 May 2021 | Accepted: 29 May 2021 | Published: 20 September 2021
Keywords: stomach; gastritis; autoimmune atrophic pangastritis; autoimmune gastritis
Atrophic gastritis can be environmental in origin and involve the antrum or autoimmune in origin and involve the body and fundus. We present a rare case of autoimmune atrophic pangastritis (AIAP), a distinct type of autoimmune gastritis (AIG) affecting the entire stomach, which should be considered in patients with other autoimmune disorders.
Atrophic gastritis is categorized as: 1) autoimmune (formerly type A) or 2) environmental (formerly type B, most frequently from H. pylori infection), with the latter being the most common. [1] These are classified by location, histology, and etiology. AIG inflames the stomach body, fundus and spares the antrum while environmental gastritis inflames the antrum. [2] Histologically, AIG involves autoimmune destruction of oxyntic mucosa, and subsequent neuroendocrine (enterochromaffin-like cell) hyperplasia from the hypergastrinemia due to parietal cell loss[3,4].
AIAP, a rare type of AIG, has been described in only 15 cases since being formally recognized in 2006.1 Risk factors include autoimmune and connective tissue disease with females slightly more at risk. Clinical presentations vary widely, from nonspecific symptoms of nausea, vomiting and abdominal pain to diarrhea, fat malabsorption and malnutrition. Endoscopically, patients present across a spectrum -from normal appearing mucosa to multiple gastric ulcers [5]. Therefore, diagnosis relies on histology and is characterized by: 1) intense mucosal lymphoplasmacytic inflammatory infiltrates, persisting even to severe glandular atrophy, 2) pangastric distribution, 3) lack of H.pylori, and 4) lack of neuroendocrine hyperplasia.
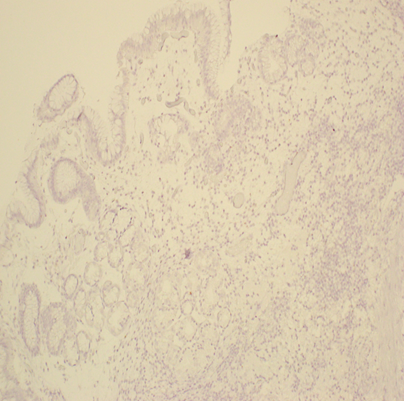
A 76 year old woman with hypothyroidism (unknown cause), rosacea, and recurrent basal cell carcinoma presented in 2013 for further management of AIAP, which was initially diagnosed by her private gastroenterologist in 2012 after 2 months of persistent nausea and vomiting with a 10 pound weight loss. Endoscopy revealed erythema, congestion, and friability in the entire stomach (Figure A). Initial biopsies by her private gastroenterologist were confirmed by Johns Hopkins University’s pathologists. Biopsies from the antrum revealed chronic inflammation with chronic focal active gastritis (Figure B). Gastric body biopsies also showed chronic inflammation with glandular dropout (Figure C). No H. pylori or neuroendocrine hyperplasia was identified on multiple biopsies with H&E staining and immunohistochemistry (Figure D). Given a 95% sensitivity and 98% specificity for diagnosing H.pylori by histology, further testing for infection was not pursued. Lab work did not detect intrinsic factor antibodies.
The patient was started on prednisone and low dose azathioprine (initiated after her basal cell carcinoma diagnosis) and she responded with improved appetite and weight gain. Prednisone was tapered off and she continued azathioprine monotherapy but held it from 2013 to 2014 due to recurrent basal cell carcinoma. She received numerous excisions (last one in 2017) for her basal cell carcinoma including on her cheek, arms and back. She is currently in remission and continues to follow dermatology. She was later diagnosed with breast cancer in December 2015, underwent right mastectomy in 2016, and completed chemotherapy and radiotherapy by November 2016. The patient’s azathioprine was reduced during this time and she complained of nausea and vomiting that is unclear if it was related to AIAP or from chemotherapy. Azathioprine was continued intermittently for four years total before weaning off due to long-standing clinical remission. The patient’s AIAP remains in clinical remission off all therapy after two years. We plan to repeat an upper endoscopy this year.
Our patient’s atrophic gastritis with diffuse atrophy throughout the stomach, full-thickness mucosal inflammatory infiltrates not related to H.pylori infection, and lack of neuroendocrine hyperplasia are consistent with AIAP, a distinct form of AIG with unclear etiology.
A case series of 8 patients showed a slight female predominance, systemic autoimmune and/or connective tissue disease in all, and autoimmune markers in 7 of 8 patients. Given its strong association with systemic autoimmune disease and the development of low-grade dysplasia in 1 patient, Jevremovic et al. suggested an autoimmune etiology against multiple cell lineages with subsequent neoplastic potential. As much as 10% of AIG cases may progress to adenocarcinoma or carcinoid tumor. In contrast, the lack of neuroendocrine hyperplasia in AIAP suggests lower risk of endocrine tumors, but the risk of adenocarcinoma persists [5,6].
Autoimmune diseases like lupus, Type I diabetes, and vitiligo may all precede or follow the diagnosis of AIAP, similar to AIG [7]. Interestingly, our patient was noted to have a history of hypothyroidism of unknown etiology that is currently well-controlled on levothyroxine. Interestingly, up to 40% of those with Hashimoto’s thyroiditis have gastric disorders and Hashimoto’s is present in about 40% of patients with autoimmune atrophic gastritis.[8] Rosacea, present in our patient, may also have an autoimmune component, and is associated with multiple immune mediated gastrointestinal disorders such as celiac disease and inflammatory bowel disease.9In patients with concomitant autoimmune diseases undergoing upper endoscopy, the finding of an autoimmune disorder should raise suspicion for the possibility of AIAP.
Our patient had a negative intrinsic factor antibody but the use of traditional serum antibody testing for AIG is limited by low sensitivity and specificity.5 The ratio of pepsinogen 1 and 2, however, reflects the gastric mucosal morphologic state and may serve as a marker of pangastritis, since pepsinogen 1 decreases as superficial gastritis progresses to atrophy.[11]
The treatment of AIAP centers around immunosuppression. Given only a handful of case reports of AIAP, there is scant data regarding the best treatment modalities and clinical outcomes in adults. Most cases describe treatment with prednisone and/or azathioprine in pediatric patients. Symptoms most often resolve with treatment; however, on follow-up biopsies after treatment, abnormal findings of inflammation and dysplasia may persist for months, which support the risk for adenocarcinoma. Unlike AIG, there are no current guidelines in terms of treating, screening, and monitoring patients with AIAP and further investigation is warranted.[12,13,14]
Figure Legend:



Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
