
Case report | DOI: https://doi.org/10.31579/2578-8868/296
1 The University of Chicago Medical Center, Department of Neurology, Chicago, IL
2 Spartan Health Sciences University School of Medicine, Department of Neurology, Vieux Fort, LCA
3 American University of Integrative Sciences School of Medicine, Barbados
4 International University of the Health Sciences Medical School, Basseterre, Saint Kitts and Nevis
5 Stanford University, Cardiovascular Intensive Care Unit, Stanford, CA
*Corresponding Author: Asia Filatov, The University of Chicago Medical Center, Department of Neurology, Chicago, IL
Citation: Asia Filatov, Tirtha Sawant, Andrew Hariraj, David Whitfield and Louisa Chan et al, (2024), Statin Myopathy: A Case series, J. Neuroscience and Neurological Surgery, 15(1); DOI:10.31579/2578-8868/296
Copyright: © 2024, Asia Filatov. This is an open-access article distributed under the terms of The Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Received: 28 December 2023 | Accepted: 23 January 2024 | Published: 30 January 2024
Keywords: statins; necrotizing myopathy; hmg-coa reductase inhibitors; immune-mediated necrotizing myopathy
Anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) immune-mediated necrotizing myopathy (anti-HMGCR IMNM) is a rare adverse effect associated with the use of statin medications [1]. While the investigation remains ongoing, statin exposure is thought to induce mitochondrial dysfunction due to inhibition of HMGCR, subsequently triggering an inflammatory cascade with oxidative stress and autoantibodies (aAbs) against HMGCR, ultimately leading to an immune-mediated necrotizing myopathy (IMNM) [2]. Clinical presentation may involve dermatomyositis-like features ranging from myalgias to severe and progressive proximal muscle weakness, often persisting after cessation of the offending statin [3]. Serum studies typically reveal significantly elevated Creatine Kinase (CK) levels, often ranging from 10 to 100 times the upper limit of normal [4]. Presumptive diagnosis is confirmed via muscle biopsy, which typically reveals necrotic fibers with minimal inflammatory cell infiltrates [5]. In addition to withdrawal of the offending agent, treatment may involve immunomodulatory medications (e.g. intravenous immunoglobulin (IVIG), steroids, and/or Rituximab). For most patients, resolution typically occurs over months, sometimes years. This paper highlights three cases of statin-induced myopathy and the clinical implications for providers in prompt recognition, diagnosis, and management of this rare condition. Further research is needed to better understand both the risk factors and underlying mechanisms in order to increase awareness, optimize care, and minimize the morbidity associated with this adverse drug reaction.
A 75-year-old male with a past medical history (PMH) of hypothyroidism, prostate cancer (in remission), and squamous cell carcinoma of the lung presented for evaluation of muscle weakness. He was recently admitted for encephalopathy and rhabdomyolysis in the setting of one year of progressive weakness. Approximately one year prior to admission, he developed proximal leg weakness, most notably while walking up stairs. Due to the insidious onset of this weakness, along with superimposed back pain, he was given a preliminary diagnosis of lumbosacral disease and underwent an electrical stimulation implant with pain management. While he reported interval improvement in back pain, weakness persisted. Six months later, he began experiencing painless weakness in the upper extremities, first with overhead activities followed by difficulty with opening jars. On the day of admission, he became acutely confused. A stroke alert was initiated for acute altered mental status (AMS); National Institutes of Health Stroke Scale (NIHSS) was zero, however, both his responses and movements appeared to be slowed. Remarkable labs include CK > 4000 and normal ESR/CRP. He confirmed taking Lipitor for several years. Due to concern for statin-induced myopathy, intravenous (IV) fluids in conjunction with a 3-day course of high-dose IV steroids were administered. Upon day of discharge, his CK had decreased.. An MRI of the brain revealed incidental findings of chronic caudate lacunar infarct not thought to be contributing to his current symptoms. His AMS was attributed to acute, toxic metabolic encephalopathy (ATME) and he was subsequently discharged to rehab. While undergoing rehabilitation, mental status returned to baseline. However, weakness continued to progress. He then noticed a rash on his legs localized to the bilateral shins; the rash was erythematous with scaly patches. He applied cortisone cream to improve the itching. Still, the rash persisted later appearing on his hands. Otherwise, the chronic scaliness and discoloration spanning his arms, face, and scalp, which he felt were related to chronic sun exposure, were unchanged. He complained of progressive difficulties with balance. Muscle biopsy revealed necrotic muscle fibers and an absence of inflammatory infiltrates consistent with a diagnosis of anti-HMG-CoA reductase antibody-associated myopathy. The diagnosis was established; the patient had discontinued statin on previous hospitalization and was now concurrently treated with prednisone 60mg and azathioprine.
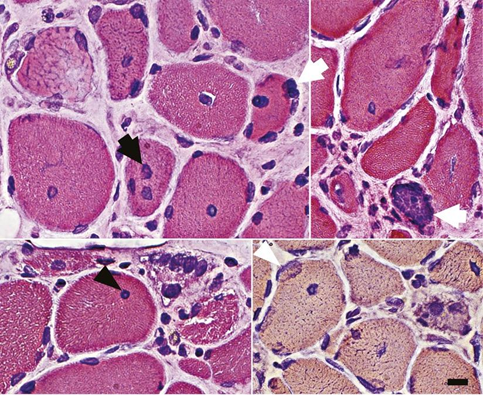
Figure 1: Abnormal myofiber nuclei in anti-HMG-CoA reductase antibody–associated myopathy.
An 84-year-old male with PMH of hypertension, hyperlipidemia, pulmonary embolism, and type 2 diabetes mellitus presented to the hospital with a complaint of bilateral lower extremity weakness. He was able to stand from a sitting position without assistance. He had trouble swallowing solid foods; otherwise, without complaints. He was last seen at our institution October 2022 for left frontoparietal subdural hematoma and bilateral pulmonary emboli (PE). He was started on warfarin at that time. A month later, he was readmitted for reported loss of consciousness and worsening bilateral lower extremity weakness; at that time, subdural hematoma and bilateral PE had resolved. Since December 2022, he had been using a walker for assistance with balance due to worsening gait and difficulty climbing into the van. Serum studies showed persistently elevated CK despite aggressive hydration. Electromyography (EMG) and nerve conduction studies (NCS) were performed for bilateral lower extremity weakness with findings suggestive of a myopathy affecting the bilateral iliopsoas muscles. Other myopathic units seen were in the right deltoid and flexor carpi radialis muscles. An MRI of the brain showed no new infarcts or subdural hematoma and no evidence of cervical spinal stenosis. A muscle biopsy was performed which confirmed the diagnosis of necrotizing myopathy. He was subsequently started on high-dose steroids for 3 days. HMG-CoA reductase antibody level was 109 (>20 is considered a positive predictive biomarker; < 20>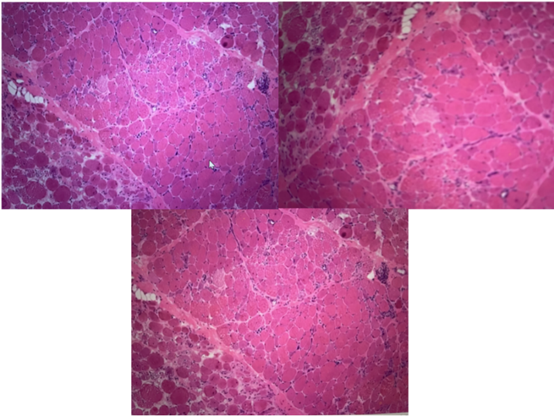
Figure 2. Muscle Biopsy Findings.
A 68-year-old male with PMH of hypertension, type 2 diabetes mellitus, and hyperlipidemia (on atorvastatin) presented from a skilled nursing facility to the general neurology clinic via primary care referral for inability to walk. His difficulties began with proximal left leg pain which extended to the anterior left thigh and subsequently the right leg, with concomitant, progressive weakness in the arms and legs. He gradually started limping. Approximately six months prior to presentation, he lost the ability to ambulate due to pain and weakness in his legs. He was wheelchair bound. EMG/NCS revealed positive sharp waves and fibrillation potentials with and without myotonic discharges and small amplitude motor unit action potential (MUAP) recruitment in select muscles of the right upper and lower extremities consistent with an inflammatory myopathy. Serum CK was elevated at >7000. A biopsy was performed to confirm suspicion of statin-induced myopathy (Figure 3). He was accordingly admitted to the inpatient general neurology service and received two days of intravenous immunoglobulin (IVIG) therapy. Additionally, he completed a 3-day course of IV high-dose steroids and methotrexate with instructions to continue a steroid taper upon discharge.
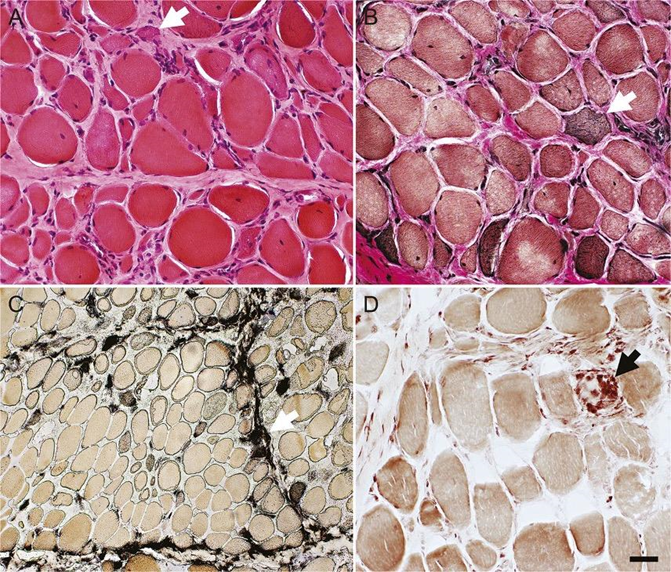
Figures
In this case series we describe three cases of statin-induced HMGCR IMNM. While it has been observed that around 20% of individuals exposed to statins experience muscle symptoms [6], this is often attributed to direct statin-induced toxicity [7] rather than anti-HMGCR autoantibodies [8]. The majority of patients with anti-HMGCR IMNM experience persistent myopathy with elevated CK levels following statin discontinuation with improvement only following initiation of immunomodulatory therapy [9], which may include IVIG, steroids, and Rituximab [10, 11]. In contrast to other idiopathic inflammatory myopathies (IIM), such as anti-signal recognition particle (SRP) myopathy, anti-HMGCR IMNM is uniquely associated with statin exposure [10]. While the pathophysiology remains unclear, current understanding posits that statin exposure may initiate an autoimmune cascade by first increasing the expression of autoantigens. These autoantigens then result in upregulation of HMGCR (which statins typically inhibit) in regenerating muscle cells and resting myocytes [12]. This autoimmune response is then perpetuated via a persistent self-antigen [12].
The incidence of IIM overall is estimated to be 0.2 to 2 per 100,000 person-years [13]. Among IIMs, the exact prevalence and incidence of statin-associated IMNM is unknown. However, one study in the United States estimates the prevalence at 7-11 per 100,000 people [10]. Another publication estimates that IMNM comprises approx. 6-10% of all IMMs [14]. Anti-HMGCR IMNM has been frequently observed among women over the age of 40 [14]. In the PRIMO study, it was found that anti-HMGCR IMNM risk was associated with dose-response effect: patients in the study taking Atorvastatin 10mg had 12.5% risk of anti-HMGCR, 21.2% with atorvastatin 20mg, and 28.4% with atorvastatin 40mg [15]. Additional risk factors for acquiring anti-HMGCR IMNM included advanced age (≥80 years of age), multi-system disease, kidney disease, untreated hypothyroidism, drug-drug interactions, excessive alcohol intake, major surgery, trauma, diet, and genetics [16]. As in the cases described above, progressive muscle weakness and soreness are among the most common presenting signs and symptoms. When confronted with these symptoms in the setting of statin use, clinicians should entertain a broad differential including but not limited to consideration of common myopathies secondary to thyroid disorders, paraneoplastic disorders, polymyositis, dermatomyositis, and inclusion-body myositis [17]. Serum levels of CK and thyroid-stimulating hormone (TSH) are helpful; if CK levels are elevated 4-10 times the upper limit of normal, renal function panel and urine myoglobulin markers should be obtained to rule out rhabdomyolysis [18]. Individuals should then undergo antibody testing to assess for the presence of anti-HMGCR antibodies, which are strongly associated with statin-induced anti-HMGCR IMNM [19]. This can additionally help distinguish this condition from other causes of autoimmune necrotizing myopathy, including antibody-negative IMNM and anti-signal recognition particle (SRP) myopathy [13]. MRI, while not necessary, may show contrast enhancement and muscle edema in areas of localized inflammation [20]. While not required for diagnosis, muscle biopsy typically reveals necrotic muscle fibers with varying degrees of minimal leukocyte infiltration [21]. A notable feature in all of our cases was prolonged symptoms beyond statin cessation. This is characteristic of anti-HMGCR IMNM, where some studies have demonstrated the persistence of necrotic fiber infiltration on serial biopsies performed months to years following the onset of symptoms [12]. First and foremost, the management of suspected anti-HMGCR IMNM consists of removal of the offending agent. Most patients will then require some form of immunomodulatory therapy; commonly steroids and/or IVIG but also other drugs including methotrexate, azathioprine, mycophenolate mofetil, or rituximab [20]. IVIG may be re-considered in refractory cases, with earlier administration associated with improved outcomes in some case studies [22]. Coenzyme Q10 (CoQ10) supplementation demonstrates no significant benefit in improving symptoms [23]. In addition to the above, clinicians should focus on supportive care, including pain management and physical therapy. Alternative statins or non-statin lipid-lowering agents (e.g. evolocumab) may be considered in patients who require cholesterol-lowering therapy.
Anti-HMGCR IMNM is a rare IMNM associated with the use of statins. The typical clinical presentation involves myalgias and progressive proximal muscular weakness, which often persists for months to years following statin discontinuation. Lab work may reveal elevated serum CK levels 10-100 times the upper limit of normal. While not required, muscle biopsy is often used to confirm diagnosis. Treatment involves discontinuing offending statin, often followed by administration of immunomodulatory medications. To date, there are no standardized or validated guidelines for management of this condition. Further research is necessary to identify best practices for diagnosing and managing this condition.
In the most recent reviews on treatment for AA [133], psoriasis [134], and vitiligo [135]TNF-α, IL-6, IL-1β, and IFN-γ have been targets of immunotherapy. These targets may be more comprehensively addressed without toxic side effects by addressing the gut dysbiosis associated with these autoimmune diseases. A regimen of prebiotics, probiotics, postbiotics, vitamin D, and magnesium is at the top of this approach (see figure 2). This novel therapeutic approach to autoimmune skin diseases outlined in this review is in part theoretical and awaits an appropriately structured randomized controlled trial on the efficacy of d-mannose and butyrate in this regard. It may enhance or possibly replace immunotherapy in some who are so afflicted. Relief would flow to women (LC/T1DM87/other autoimmune diseases), especially those of color (AA [136], psoriasis [137], vitiligo [138], AD [139], T2DM [140]).
None
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
