
Research Article | DOI: https://doi.org/10.31579/2693-7247/001
Department of Physics, A.V.C. College, Mayiladuthurai, Tamilnadu, India.
*Corresponding Author: S. Ramalingam, b Department of Physics, A.V.C. College, Mayiladuthurai, Tamilnadu, India.
Citation: S. Ramalingam (2018) Spectroscopic and computational (DFT-SCF) investigations on anti tuberculosis activity of Isonicotino hydrazide .J Pharmaceutics and Pharmacology Research 1(1) DOI: 10.31579/2693-7247/001
Copyright: © 2018 S. Ramalingam This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 19 January 2018 | Accepted: 14 February 2018 | Published: 23 February 2018
Keywords: isonicotino hydrazide, σ-bond, δ-bond, NMBO, anti tuberculosis, Polarizability, phermodynamic activity.
The molecular spectroscopic investigations have been carried out to study the antibiotic activity of the Isonicotino hydrazide. The activity of the compositional parts was tested by fundamental modes of vibrations of various bonds of the molecule. The chromophores action for the inducement of the antibiotic activity of the compound was analyzed from the electronic excitation absorption peaks. The σ-bond, π-bond and δ-bond interaction lobes were identified and the energy exchange between the orbitals was investigated from frontier molecular orbital profile. The asymmetrical charge distribution among different entities of the molecule for the perseverance of anti tuberculosis mechanism was recognized. The NMBO interaction energy transition outline was organized by the NBO calculation adapted with Gaussian calculations and the exchange of maximum energy transaction among various functional groups for the incentive of antibiotic were determined. The second order Polarizability of the compound emphasized the consistency of the antibiotic activity of the molecule. Thermodynamic activity of the molecule with respect to the temperature was stressed the decomposition rate and Gibbs free energy helped to determine the steadiness of the compound. The inhibition catalytic efficiency of the title molecule was fully tested by molecular docking study.
Isonicotino hydrazide is also named as Isoniazid, is an antibiotic compound used as a first-line agent for the prevention and treatment of both latent and active tuberculosis [1]. It is effective organic compound against mycobacteria, particularly Mycobacterium tuberculosis. It is also active against some atypical types of mycobacteria [2]. Isonicotino hydrazide is available in tablet, syrup, and injectable forms [3-4]. It was considered as a starting point in the search for new active derivatives and analogues such as hydrazones which have been reported as active anti-TB drugs [5-6]. Isonicotino hydrazide is a pro drug and must be activated by bacterial catalyze. Specifically, activation is associated with reduction of the mycobacterial ferric Kat G catalase- peroxidase by hydrazine and reaction with oxygen to form an oxyferrous enzyme complex. Once activated, Isonicotino hydrazide inhibits the synthesis of mycoloic acids, an essential component of the bacterial cell wall. At therapeutic levels isoniazid is bacteriocidal against actively growing intracellular and extracellular Mycobacterium tuberculosis organisms.
Isoniazid is an important component used in “triple therapy” to combat tuberculosis. It has reduced Tabletting formulations stability. Anti-oxidants are obligatory to counter oxidative stress, pulmonary inflammation, and free radical burst from macrophages caused in tuberculosis and other diseases. Isoniazid has long been used clinically to a great extent as the primary drug in the treatment and prophylaxis of tuberculosis. This drug has been considered to be one of the potent sensitizing drugs presumably capable of being covalently incorporated into a variety of protein acceptors in vivo and causing a wide variety of hypersensitivity symptoms as a result of autoimmunization.
Isoniazid has a simple organic structure which containing two essential components required for the high activity against Mycobacterium tuberculosis, i.e., a pyridine ring and a hydrazide group [7-8].Isoniazid inhibits the synthesis of mycolic acids, an essential component of the bacterial cell wall. At therapeutic levels isoniazid is bactericidal against actively growing intracellular and extracellular MTB organisms. Isoniazid is used in conjunction with other effective anti-tuberculosis agents under multi-drug therapy [9].
After screening the available literature, it is found that, there is no work has been carried out to analyze the root cause of the anti tuberculosis activity of the compound using Spectroscopical tool supported by computational calculations. In the present study, the FT-IR, FT-Raman, NMR and UV-Visible spectral investigations have been carried out is to rendering the pharmaceutical activity.
Physical state:
Recording profile:
Using Gauss view, the present molecule was designed with optimized geometry and such geometry was used to run the program. The entire calculations were performed to calculate all the parameters which were used to analyze in different aspect. The composite molecule made by base and substitutional combinations where the change of geometrical parameters, Mulliken charge levels and the vibrational spectral properties were tabulated for the modification of physical and chemical parameters. The entire quantum chemical computations were performed using the Gaussian 09 D. 01.version software program in core i7 computer [13].
The computational calculations were performed for determining unknown geometrical parameters, vibrational frequencies, simulation of molecular structure and spectra using B3LYP and B3PW91 methods adopted with 6-31++G(d, p) and 6-311++G(d,p) basis sets. The energy absorbance by the present compound related with electronic spectra, the NBO and HOMO-LUMO energies were calculated using time-dependent SCF method with best fit basis set. In the same way, the 1H and 13C NMR chemical shifts with respect to TMS were calculated by GIAO method using I-PCM model in combination with B3LYP/6-311++G(d,p). The Mullikan charge assignment on different parts of the compound was calculated and was purposely elucidated for the determination of key factor for pharmaceutical activity of the compound. The dipole moment, linear Polarizability and the first order hyper-Polarizability in different coordinate system of the compound were computed using B3LYP method with the 6-311++G(d,p) basis set. The ECD and VCD spectra were simulated from available frequencies and the optical chirality was studied and the mechanism for masking the toxicity was interpreted.
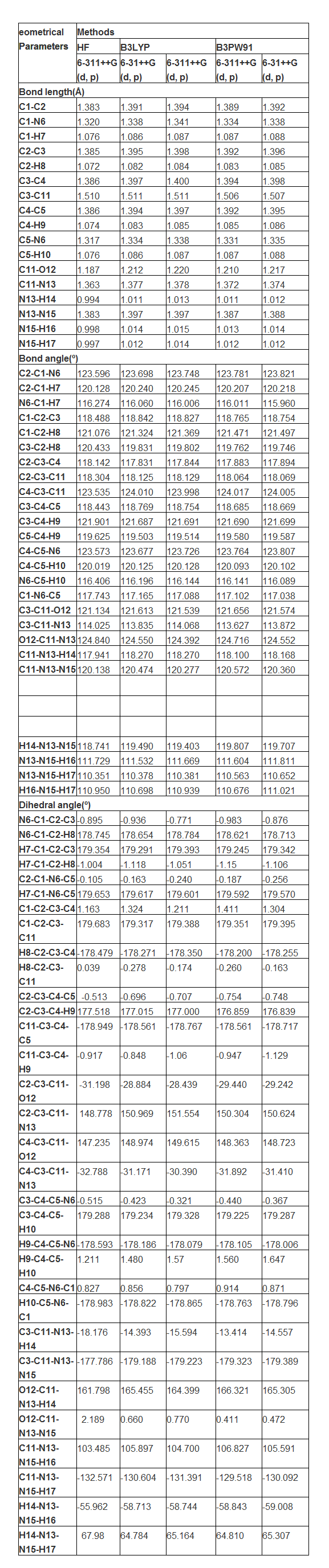
4.1. Structural Deformation analysis
The bond type of present compound was displayed in the Figure 1 with density of energy states. The present compound was made on pyridine ring and it was substituted by carbonyl and amine groups. Generally, due to the substitutions, the fundamental frame of the molecule is distorted [14]. Accordingly, in this case, the pyridine ring was found to be broken on par with the substitutions. The value of bond length of C-C and C-N of the ring without substations were C1-C2: 1.393, C1-N6:1.336, C2-C3: 1.391, C3-C4: 1.391, C4-C5: 1.393 and C5-N6: 1.336 respectively. Whereas, the value of same bond length of C-C and C-N of the ring with substitutions were C1-C2: 1.389, C1-N6:1.334, C2-C3: 1.392, C3-C4: 1.394, C4-C5: 1.392 and C5-N6: 1.331 respectively. From this order, it was found that, in the case of C1-C2 and C4-C5, the bond lengths were decreased up to 0.004 Å. But, the bond length C2-C3 and C3-C4 increased by 0.003Å due to the substitutions. In the case of C1-N6 and C5-N6, the bond length was decreased up to 0.005Å. All these asymmetric change of bond length was due to the insertion of ligand group. The impact of such physical parameter change was observed in the change of physical as well as chemical property. Accordingly, the fundamental property of the pyridine compound was antipyretic substance, but after the substitution, the present compound to be acted as an anti tuberculosis agent.
The bond angle C2-C1-N6 and C4-C5-N6 were almost same as 123.7° which was very high stretching semicircle angle in the ring due to the hetero nuclear atom. The bond angle C2-C3-C4 was squeezed much as 117.8° by the injection of chain. Form this condition, it was cleared that, the corresponding mass and energy of the chain completely transferred to the ring. So, the chemical property of the pyridine ring was completely altered with respect to the ligand group. The density of energy states diagram of the present compound was displayed in the Figure 1. From the figure, it was peculiarly observed that, the energy spectral density was intensively accumulated at the place of ligand group over the ring. The energy of unoccupied level was found at the tail end and the discrete energy levels were oriented at top end of energy spectrum. This view showed the energy transformation from ligand group to pyridine ring.
4.2. Mulliken charge dislocation analysis
The molecular orbital formation is decided by symmetrical and asymmetrical charge distribution among atoms [15]. The symmetry or asymmetry of the charge distribution plays a fundamental role in determining the chemical properties of the molecule and consequently this property of the charge distribution is used as a basis for the classification of chemical bonds [16-17]. All the chemical bonds in the compound would be formed as covalent bond between composite atoms and are to be polar and non polar. Normally, in the compound, the polar and non polar bonds are existed with respect to the substitutional groups. If the ration of polar bonds is greater, the asymmetry charges distribution taking place in the compound. Here, the dipole moment of the compound was found to be high since the ratio of hetero nuclear bonds was greater than homo nuclear bonds. The Mulliken charge levels were depicted in the Table 2 and the corresponding pictorial diagram was shown in Figures 2 and 3.
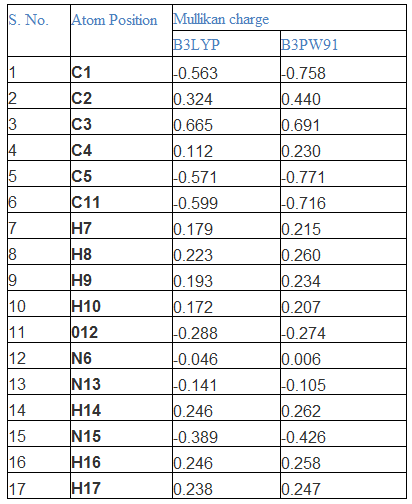
In this compound, though the hetero nuclear atom (N) present in the ring, the strong bond was created between CC of the ring and also the N was to be found as neutral. Due to the asymmetrical charge dislocation motion towards substitutional group, the top moiety of the ring becomes protonic region whereas the bottom moiety turns out to be electron rich region (depletion region). From this view, it was inferred that, the considerable amount of chemical energy was transferred from ring to ligand group. Normally, the rich polar character present in carbonyl group, but in this case, the electronic charges were accumulated vigorously and the bond come in to repulsive. On the other side of the substitutional group, in order to make strong attractive bond, the intermediate nitrogen atom converted as neutral.
4.3. Vibrational analysis
4.3.1. Vibrational assignments
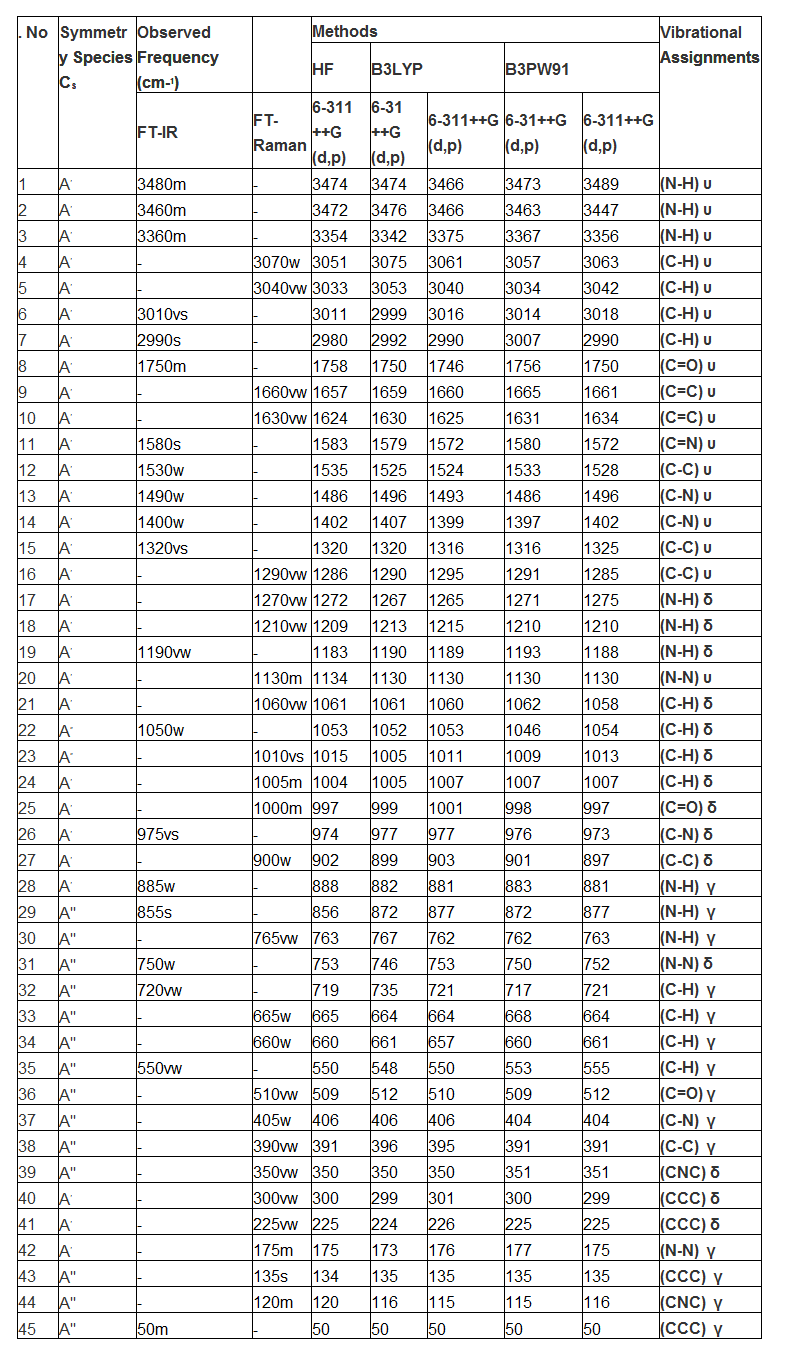
The fundamental frequency pattern of the compound was represented by IR and Raman spectra and the observed spectral peaks in terms of wavenumbers were presented in the Table 3 and the observed with simulated FT-IR and FT-Raman spectra were presented in the Figure 4&5. According to the vibrational degrees of freedom, since the present compound contained 17 atoms, there were 45 fundamental modes of vibrations were observed. Out of 45 vibrational modes, 17 stretching, 14 in plane and 14 out of plane bending modes were distinguished. Since the title molecule was non Centro-symmetry, the vibrations are active in IR and those inactive in Raman and vice versa. The total elementary vibrations were ordered as Г= 31Aʹ+ 14Aʺ
4.3.2. C-H vibrations
Normally, the ring C-H vibrations in benzene and pyridine ring ensured that, whether the hexagonal frame has substituted by ligand or not. If the observed fundamentals are observed within the expected region, the ring will not be affected much by the substitutions. Otherwise, the ring vibrations are rather suppressed and ratio of the same showed the impact of the substitutions on the ring. Here, the substitutional group was present at one C of the ring and 16 C-H vibrations are possible. Accordingly, the C-H stretching vibrations were found at 3070, 3040, 3010 and 2990 cm‑1 in IR and Raman spectra. The in plane and out of plane bending vibrations were observed at 1060, 1050, 1010 and 1005 cm‑1 and 720, 665, 660 and 650 cm‑1 respectively. The hetero aromatic C-H stretching vibrations are normally found between 3100 and 3010 cm‑1, the C–H in plane bending frequencies appear in the range of 1000 - 1310 cm−1 and C–H out of plane bending vibration in the range 750 - 1000 cm−1 [18-20]. The entire vibrational bands are expected to observe with medium to strong intensity. But all the bands were found with very week to very strong intensity. One peak of stretching and entire signals of out of plane bending were observed out of the expected region whereas all the in plane bending modes were found at the top end of the allotted region. The observed signals were affected in low energy region which was mainly by the energy transformation from ring to ligand group in order to generate anti-tuberculosis activity.
4.3.3. Ring CC and C-N vibrations
The main hexagonal frame of pyridine ring is constituted by C-C bonds and these are directly affected by the loaded ligand groups. In addition to that, the ring associated with N atom, so the ring vibrations include the C=N and C-N vibrations also. Interactions between ring C=C and C=N stretching vibrations result in two strong-to-medium intensity absorptions about 100 cm-1 apart [21]. Usually, the C=C and C=N stretching modes are observed in the region 1615-1575 cm‑1 and 1520 -1465 cm‑1 respectively [22-25]. Accordingly, in the present case, the C=C and C=N stretching signals appeared at 1660 & 1630 and 1580cm‑1 respectively. Both the stretching bands are moved well above the expected level of the spectrum. In this view, it was cleared that, the ring energy was found to be increased and which was favor for the energy transaction between ring and ligand group. Usually, these vibrations are followed by C-C and C-N stretching bands which are observed around 1460 cm-1[26]. In this case, these vibrational bands were observed at 1530 cm‑1 and 1490 & 1430 cm‑1 respectively. The CCC and CNC in plane and out of plane bending (ring breathing) modes were identified at 300, 225& 175 and 135, 120 & 50 cm‑1 respectively. These observed fundamental modes moved down to well below the expected region. The ring deformation vibrations have been restricted since it was substituted by the carbonyl and amino groups. Finally, it was concluded that, the ring energy was consumed lot for the formation of drug property.
4.3.3. NH2 vibrations
Specifically, the amino group is always dominating all other groups and it very much pronounced in the drug property of the compound [27]. In this case also, the amino group was played important role to induce such anti-tuberculosis activity. Usually, the N-H stretching modes are appeared in the region 3300-3500 cm‑1[28-29]. Therefore, the same signals for N-H stretching found with medium intensity at 3480, 3460 and 3360 cm‑1 in IR only. As in the previous case [30], such vibrations were observed at the top end of the allocated area. Form this observation, it was cleared that, these components were found to be active and it took part in transaction of energy for creating novel property.
The N-H in-plane bending vibrations (scissoring) are usually observed in the region 1610-1630cm−1 and the out of plane bending (wagging) vibrations are normally identified in the region 1150-900cm−1 [31-32]. Here, these vibrations (in plane and out of plane) have been observed at 1270, 1210 & 1190 cm‑1 and 885, 855 & 765 cm‑1 respectively. Dissimilar to the stretching, these bending modes were found to be affected and depressed much which was due to the presence of carbonyl group. The activeness of bending modes rather decreased by C=O group by inductive effect.
4.3.4. C=O, N-N and C-C vibrations
The ketones related Carbonyl group vibrations are the important characteristic bands in vibrational spectra since it acts as major part of creating complete property and therefore, these bands have been the subject of wide analysis [33-34]. Normally, the intensity of these bands would be greater since its conjugation and dipole moment. Therefore, it leads to the intensification of the scattering lines as well as very strong intensity of infrared band. The carbonyl stretching vibrations in ketones are expected in the region 1680–1715 cm−1[35]. In this case, a band was found with medium intensity at 1750 cm‑1 in IR only. The band was appeared well above the allowed region but its intensity was suppressed which was due to the nearby amino group. It’s in plane and out of plane bending vibrational modes were observed at 1000 and 405 cm‑1 respectively. These vibrational modes were agreed well with the previous work [36].
The amine groups were attached with one another by N-N bond and forming azine group and it was very significant since it is Y-junction which exchanges the energy between ring and amine group of molecule. Generally, the azine bond N-N stretching vibration was found around 925-1150 cm‑1 [37]. The N-N stretching vibration was observed with medium intensity at 1130 cm‑1. The consequent in plane and out of plane bending modes were observed at 750 and 175 cm‑1 respectively. The acquired vibrational bands were precisely harmonized with the literature and ensure the role of azine group in the drug properties of the compound.
The C-C stretching of C-C bond of out of the ring are observed in the region 1470-1520 cm‑1[33]. Here, the stretching modes have been found at 1320 and 1290 cm‑1. The corresponding in and out of plane bending vibrations was observed at 885 and 390cm‑1 respectively. These bands were at exact allowed region since it was favored by the carbonyl and amino groups.
4.4. NMR confinement
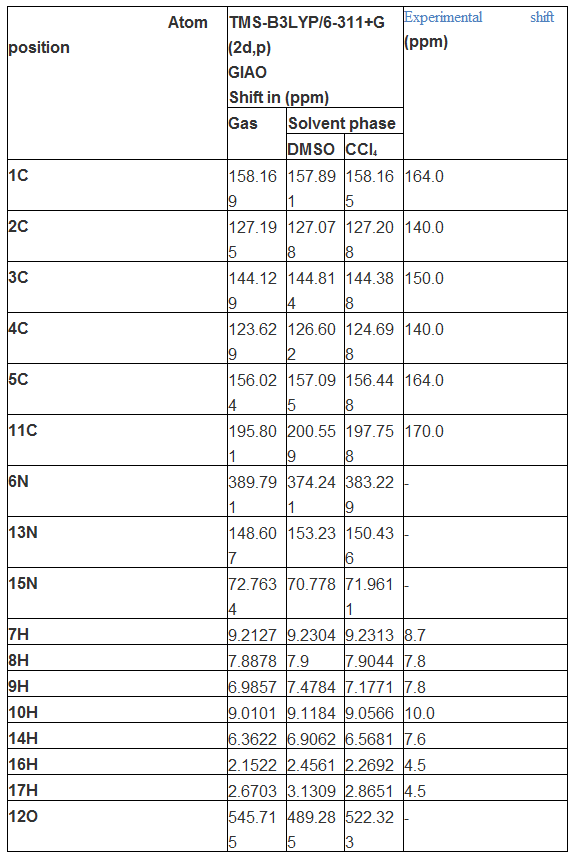
The 1H and 13C NMR spectral data was presented in Tables 4 and the corresponding spectra were shown in Figure 6. Generally, the chemical shift of carbons and hydrogens are situated in different environment with respect to the coupling constant and makes the molecule complicated. The isotropic chemical shift of C and H are usually explicated the unknown chemical properties of complicated compound. In general, the chemical shifts of carbon is depends upon the bonded atoms and their chemical environment in the compound. The isotropic chemical shifts in aromatic compounds are universally observed in the region of 100- 190 ppm [38]. The chemical shift of C of base compound with respect to ligand attachment is providing direct the dependable interpretation of chemical properties. Usually, the chemical shift of aliphatic chain is constantly behind the aromatic compound [39]. Symmetry also makes atoms equivalent in molecules and if group of atoms are in some identical locations relative to a plane of symmetry in a molecule, they will be equivalent [40-41]. Here, the carbons of the ring were located in the region 140- 160 ppm as in normal case.
Generally, the NMR signals are falling into domain with respect the diamagnetic equivalence of carbon and hydrogen atoms. The observed chemical shift of carbons of the pyridine ring; C1, C2, C3, C4 and C5 were restricted in the region 140-164 ppm. The observed chemical shift of C1 and C5 were found to be same (164ppm) which was more than rest of others due to coupling of N whereas the calculated shift was 156 and 158 ppm respectively. This condition also due to the asymmetrical electron orientation on such carbons which was evident in Mulliken charge analysis. Similarly, C2 and C4 were same and 140 ppm, but the observed and calculated chemical shift of C3 was 150 and 144 ppm respectively. The observed and calculated chemical shift of C11 was found to be 170 195 ppm respectively which is greater than others since its electron cloud was fragmented asymmetrically by ring, carbonyl and amino groups. The C3 and C11 have been opened up due to the random breaking of proton shield by π-bonded oxygen and amine groups in which the consistent amount of energy was exchanged among different entity of molecule in order to enforce the drug property in the compound.
The hydrogen atoms were attached with C and N atoms in the molecule which make different environment upon which the chemical shift of the atoms was classified. Here, the chemical shift of H in ring was observed in the region 7-10 ppm whereas in amino group, the shift was observed 4.5 and 7.6 ppm. The chemical shift was observed consistently with respect to the attached atoms.
4.5. Frontier molecular outline
The interaction between two atomic or molecular orbitals will form two new orbitals. One new orbital is antibonding orbital which has the higher energy than the original molecule orbital. The other new orbital is the bonding orbital which is lower in energy than the initial one. The stabilization of the bonding molecular orbital and destabilization of the antibonding can increase when the overlap of two orbitals increases [42]. In the molecular interaction, there are the two important orbitals interact each other. One is the highest energy occupied molecular orbital is called HOMO. The other one is the lowest energy unoccupied molecular orbital is called LUMO. When a pair of electrons filled in one of the molecule orbital and no electron occupy in the other orbital, this interaction is very stable and called filled- empty interaction.
The orbital interaction profile of title compound was presented in the Figure 7 in which different bonding interactions were seen clearly. The elevated orbital energy level was depicted in the Table 5. In the case of LUMO, there were two sets of first order π-bonding interaction found over the ring cc of opposite moiety and one σ-bonding existed on N of the ring. Apart from that, one π-bonding interaction taking place between ring C and ligand C which was also connect =O. one N-H iso surface was observed and little flavor was identified on H. usually, the π-conjugation interaction orbitals induce active biological as well as crystal property in the aromatic compound[43]. Here, the electron cloud receiving location (LUMO) was observed to be π-orbital overlapping region where the orbital was degenerative and able to consume electronic energy from HOMO.
In HOMO, the cascade of orbital overlapping of degenerate energy levels at top and bottom moiety of the ring which connect CNC and CCC with two H. peculiarly, the δ-bond orbitals found at C3 of ring C11, O12 and N13 of ligand chain. The δ-bond interaction is very rare and if present in compound, there will be stimulation of drug activity. Here, from the orbital interaction, proved the anti tuberculosis action of the present compound. In second order orbital interaction (HOMO+1 and LUMO-1), the positive and negative intersection was found and all the degenerate energy levels were interacted to make centralized donor region for supplying electronic energy.

4.6. UV-Visible absorption analysis
UV-Visible spectral analysis provides the information regarding the ligand group which causing the outstanding chemical property of the compound. Normally, the substitutional impact shifts the electronic excitation pattern (finger print region) of the base compound. The electronic excitation parameters are presented in the Table 6 and the absorption band was displayed in the Figure 8. Here the compound architected by pyridine ring attached with amino group and carbonyl group. Generally, the pyridine ring absorbance peak identified at 190 nm with 76 Kcal/mol in the UV-Visible spectrum [44]. But, by the addition of the substitutional group, the UV band was found at 331 nm with the transition energy of 86.34 Kcal/mol by crossing the energy gap of 3.74 eV. The oscillator strength of the oscillation (transition between ground and excited state) was found to be 0.0134 with higher molar absorptivity. The shift was represented as bathochromic for the higher wavelength region. Usually, the presence of chromophores such as C=O and NH2, enable intensive absorption bands at 290 and 280 nm respectively [45]. In this case, the triplet absorption bands were appeared at 331, 319 and 293 nm with effective intensity. These absorption peaks ensure the presence of such chromophores which was the reason for the inducement of the anti tuberculosis activity.
The entire transitions in the electronic HOMO and LUMO energy levels were found to be confined within the quartz UV region of the spectrum which implies that, the title compound would not be active in visible region. Because of this condition of the compound, the drug may be inert for light radiation. The UV-Visible frontier molecular excitations were belonging to n→π* class of transitions showed the strong interaction between donor (carbonyl and amine groups) and acceptor (pyridine ring). The absorption band for title compound was transparently taking place in the UV spectrum in R-band (German, radikalartig) and constantly being with therapeutic activity. In this case, the identification of absorption band in quartz-UV region predicted that, the functional group of amine and azine entities at ring was played significant role for the inducement of pharmaceutical action.
The electronic charge distribution is responsible for electronic polarization of orbitals which was induced by local electric field called ECD. The interaction of chromophores on the base compound making two fold of energy increments for transition to excited states adjust the chemical activity of the compound which can be identified in the ECD spectra. As in the Figure 8, the ECD absorption band was identified at 290 nm which was the characteristic band of C=O and NH2 groups. This band represents uniqueness effect of chemical reactivity.

4.7. Molecular electrostatic potential (MEP) maps
The molecular electrostatic potential is asymmetric charge distribution in different entities of a molecule. The molecular interaction energy was calculated by using the equation
If the charge distribution continuous over the compound, the MEP is then given by equation at any instant

The MEP is highly informative concerning the protonic and electronic charge distribution of a given molecule. It is mainly used for asymmetric charge orientation among different atoms causing strong electrostatic potential between two extreme charge levels which is favor for the inducement of the particular biological and pharmaceutical properties. The framed Molecular electrostatic potential view was presented in Figure 9 in which the entire molecular interactive zones can be viewed. The static potential energy interaction generated between proton and electron fields is ranged from high frequency (blue) region to higher wavelength (red) region which designated that, the charge dislocation between electrophilic and nucleophilic boundaries. In a molecule, charges are polarized rigorously in negative and positive terminals, and there are significant differences in electron and proton density due to the injection of substitution in base compound. The rich electro negativity region was indicated by wealthy red and positive (proton region) almost was indicated by blue.
The two extreme ends of charge distribution was represented by the color code in the range between -7.66 a.u. (genuine red faded to yellow) to 7.66 a.u. (genuine blue faded to green). Generally, the negative region is mainly localized over the portion of highly electronegative atoms.
In this case, the electron bustle zone was captured over the O of carbonyl group, N of amino group and the N of pyridine ring. The moderate negative region was concealed over the pyridine ring and further decayed when moved towards H of ring. The protonic content was confined on the hydrogen zones over the amino group. It was abundant in around the edge of amino group and deficient in carbon bonded H. This well defined atmosphere was induced by hetero nuclear bond in ring and disorder on amino groups. Due to the electron pulling away from the ring, the electrostatic energy was found to be uniform at the centre part of the ring and acted as depletion energy grid. In each and every molecule, the number of homo and hetero nuclear bonds due to the strong ligand is root cause of the major property of the compound. In this case, the strong electrophilic-nucleophilic dipole was found between ring Carbonyl and amino groups. The out of plane ligand usually making strong receptor activity when docking is made. Here, carbonyl and amino group chain appeared as out of plane ligand which will be acting as good bridging action.
4.8. Chemical properties
The transition energy of Frontier molecular energy levels represented chemical properties and molecular reactivity descriptors of composite structure. The entire chemical parameters were presented in the Table 7. The asymmetric charge distribution was measured by the resultant dipole moment of the compound and it was found to be 4.70 and 3.97 dyne in IR and UV-Visible region respectively. The resultant dipole moment of the pyridine ring is found to be 2.64 dyne whereas the present compound was 2.06 and 1.33 dyne greater than pyridine ring which was due to the addition of functional group in the compound. This increment of dipole moment was ensured the charge depletion in the molecule where the pharmaceutical activity was induced much.
The energy gap of the frontier molecular orbitals measured usually, the chemical stability of the compound and was determined to be 2.71 and 2.31 eV in IR and UV-Visible region respectively. Both the values showed reasonable chemical stability and the compound would not be disintegrated by other toxic radiation. The electron affinity of the molecule is very important for the determination of the reaction ability of receptor protein and was found to be 7.283 which were elevated to the extreme and the reaction capability of the present compound is energetic.
The ionization potential of the compound is significant to evaluate consistency chemical-bond. The ionization potential was found to be 1.862 which was moderate and enough to maintain the chemical bond stability and molecular property. Generally, the chemical hardness is a scale of obstacle for transformation of charge whereas the electronegativity is measure of the tendency to attract electrons by inter-chemical bond. Here, both parameters were found to be 2.71 which was very low and able to have good reactive character and it was possible to add further add or alternate drug properties.
The exchange of energy between frontier molecular orbitals can be measured by electrophilicity index and is an indicator high degree of electron cloud. In this case, the electrophilicity index was recognized to be 3.857 eV, but the same was 3.05 eV for pyridine ring. The derived energy was high when compared with base ring which was due to the mono substitution of ligand groups. From this point of view, it was clear that, the maximum energy exchanged between ligand via ring for creating the antibiotic activity.
Here, the pyridine ring was the base compound and it was substituted by carbonyl and amino groups where the electrophilicity charge transfer of the compound was found to be + 1.687 which emphasized the maximum charge flow from ligand to ring. This also major cause of the present compound is an anti pyritic agent.
| Parameter | IR region | UV-Visible region | Electrophilicity charge transfer (ECT) (ΔNmax)A-(ΔNmax)B |
| Etotal (Hartree) | -472.435 | -472.273 | |
| EHOMO (eV) | 7.283 | 7.019 | |
| ELUMO (eV) | 1.862 | 2.387 | |
| DEHOMO-LUMO gap (eV) | 5.420 | 4.632 | |
| EHOMO-1 (eV) | 7.572 | 7.373 | -1.687 |
| ELUMO+1 (eV) | 1.035 | 1.274 | |
| DEHOMO-1-LUMO+1 gap (eV) | 6.536 | 6.099 | |
| Chemical hardness (h) | 2.710 | 2.316 | |
| Electronegativity (χ) | -4.572 | -4.703 | |
| Chemical potential (μ) | -4.572 | -4.703 | |
| Chemical softness(S) | 0.184 | 0.215 | |
| Electrophilicity index (ω) | 3.857 | 4.775 | |
| Dipole moment | 4.706 | 3.978 | |
Table 7: Calculated energies, chemical hardness, electro negativity, Chemical potential,
Electrophilicity index of Isonicotino hydrazide in UV-Visible region
4.9. NBMO analysis
The electron cloud transition among non bonding molecular orbital abundantly involved in the inducement of the molecular property of composite molecule. The electron charge dislocation is purposively made by exchange of potential in terms of electronic energy among different entity of the composite for the development of particular drug activity. Such energy is laid down between lower energy of bonding orbital and higher energy of antibonding orbital [46]. The entire chemical property is depends upon the electronic potential is utilized for dislocating the electronic cloud for creating unique activity of the compound. Here, the non bonding molecular orbitals were urbanized by combining base and ligand groups. It was very important to identify the exchange of energy from which identity to which identity in the molecule.
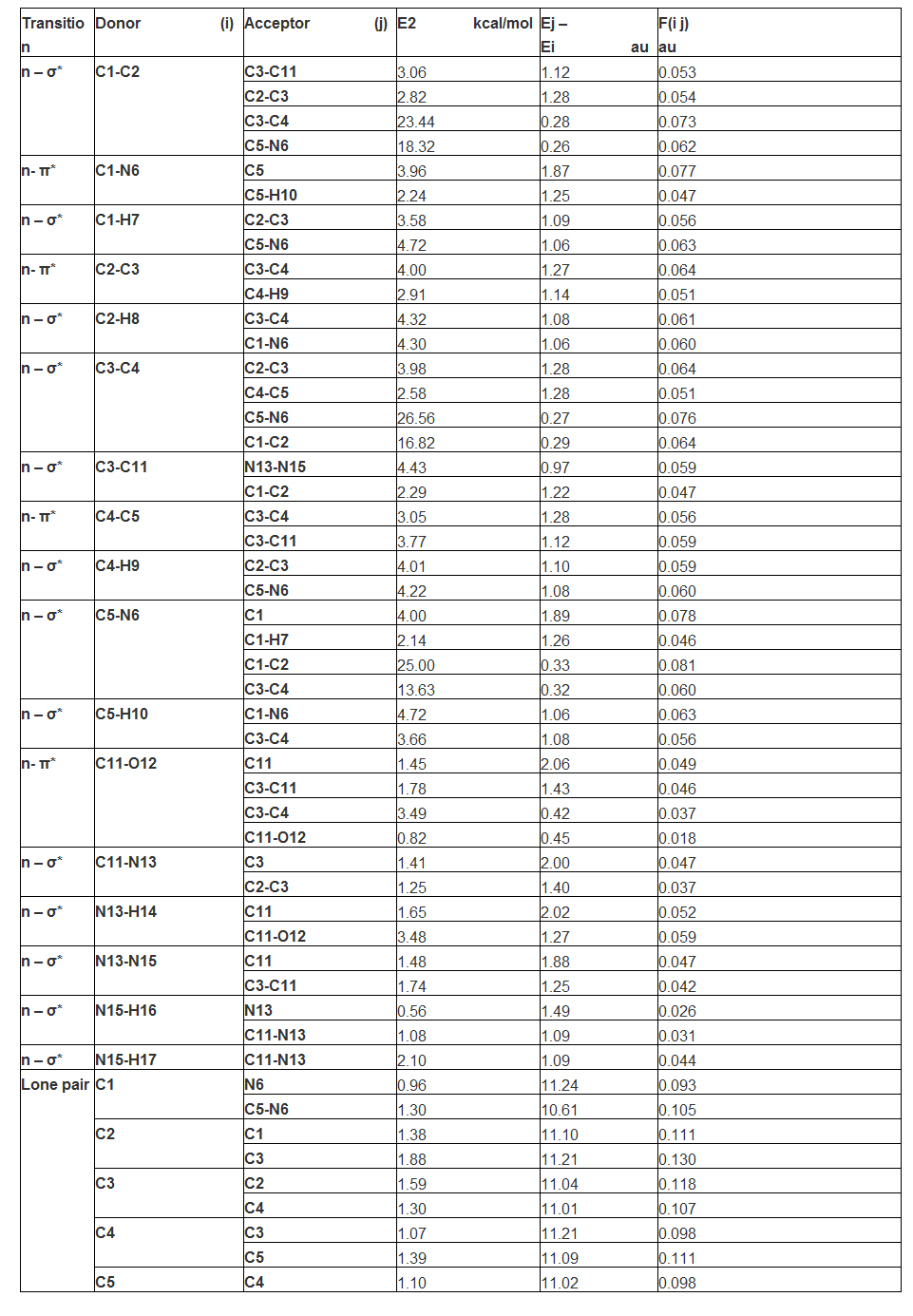
Here, in ring system, the transition from NMBO of C1-C2 to C3-C11, C2-C3, C3-C4 and C5-N6 and they assigned to n-σ* in which 3.06, 2.82, 23.44 and 18.32 kcal/mol energy respectively were transferred from first ring to ring and chain in order to connect the major ligand groups. Among those transitions, within the ring, the maximum energy was transferred. Similarly, the n-π* transition was found from C1-N6 to C5 and C5-H10 within the ring system with 3.96 and 2.24 kcal/mol amount of energy. Here, such feeble amount of potential energy was utilized to stabilize charge level in ring system. The 4.43 and 2.29 kcal/mol amount of energy
used for n-σ* transitions from C3-C11 to N13-N15 and C1-C2. The reversible transition energy from ring was directed towards ligand for stabilizing the chemical property of the compound.
In same way, the transitions assigned to n-σ* from C3-C4 to C5-N6 and C1-C2 with utilization of 26.56 and 16.82 kcal/mol energy respectively. When compared with previous, it was very potential energy was consumed for retaining the charge levels within the ring system. The transition was assigned to n- π* from C11-O12 to C3-C11, C3-C4 and C11-O12 with the respected energy of 1.78, 3.49 and 0.82 kcal/mol within the ligand group. This energy was found to be very low which was utilized to stabilize the orbitals within the ligand system. From these transitions, it was cleared that, the considerable amount of energy was exchanged within the ring system, from ring to ligand and from ligand to ring system. When compared all the transitions, the maximum amount of energy utilized to transfer the electronic cloud within the ring system for inducing antibiotic property.
4.10. Polarizability and hyperpolarizability profile
The polarized and hyper polarized orbitals are reconstituted with respect to the existence of chemical force of attraction for the molecular stabilization make possible to induce the drug property and they can be measured by calculating the Polarizability and first order hyperpolarizability tensor as in the Table 9.
The calculated value of the dipole moment was found to be very high (4.76 Debye) since the number of hetero dipole pole moments were found to be higher order and it is enough to induce first and second order polarization. The major core of ligand groups were observed on the plane of molecule and results intensive polarization. The calculated showed that, the major entities were found to be on z coordinate of the compound which point out the arrangement of ligand chain. The calculated multi pole constants in different coordinates were tabulated among which the rich polarization constants were found (~55 a.u) in xx, yy and zz coordinates. This was the main cause of the obtaining the polarization constants with considerable amount. The calculated average Polarizability and anisotropy of the Polarizability was 126.48 x10-30esu and 175.23 x10−30esu, respectively. These values were so high and to be behind to set up strong bridge between ligand and ring group which stabilize the particular drug property.
Such high values also cause of the hyper polarization action and the hyperpolarizabilityb is one of the important key factors of strong molecular orbital interaction system which was the main source of the inducement of antibiotic activity. The B3LYP/6-311++G(d,p) calculated first hyperpolarizability value (b) is 790.26 x10−33esu. From this high value of result, it was clear that, the hyper asymmetrical polarization was empowered the stabilization of frontier molecular orbitals interaction for the stimulation of therapeutic activity.
| Parameter | B3LYP6-31 | B3LYP6-311 | Parameter | B3LYP6-31 | B3LYP6-311 |
| αxx | -62.2366 | -62.2202 | βxxx | 83.0242 | 82.2134 |
| αxy | -13.2356 | -13.0668 | βxxy | -23.1768 | -22.6226 |
| αyy | -54.5181 | -54.4756 | βxyy | -6.5732 | -6.5914 |
| αxz | 5.8565 | 5.6956 | βyyy | -11.8097 | -11.7657 |
| αyz | 2.7749 | 2.8541 | βxxz | 16.0431 | 15.5269 |
| αzz | -58.7065 | -58.6774 | βxyz | -0.2716 | -0.2670 |
| αtot | 126.489 | 126.343 | βyyz | 3.5057 | 3.4570 |
| Δα | 175.234 | 175.180 | βxzz | 13.7065 | 13.6889 |
| μx | 1.7538 | 1.7653 | βyzz | -1.3945 | -1.4333 |
| μy | -3.9371 | -3.8851 | βzzz | 2.1363 | 2.0861 |
| μz | 2.0368 | 1.9839 | β | 790.260 | 790.903 |
| μ | 4.7671 | 4.7060 |
Table 9: The dipole moments µ (D), the polarizability α(a.u.), the average polarizability αo (esu), the anisotropy of the polarizability Δα (esu), and the first hyperpolarizability β(esu) of the compound; Isonicotino hydrazide
4.10. Thermodynamic function analysis
Normally, the thermo dynamical analysis on aromatic compound is very important since they offer the important information regarding the thermodynamic character of chemical reactivity. [45]. Thermodynamic functional parameters were depicted in the Table 10. The calculated entropy, specific heat capacity and enthalpy were found to be varied positively with temperature. When the temperature increased from 100K to absolute temperature 298.15, the functional parameters were varied unhurriedly whereas from 350 to 1000K, the thermodynamical functions recognized to be linear pattern and rather constant at maximum temperature. This view of variation showed the consistent chemical reactivity and considerable chemical hardness of the present compound. The Gibbs free energy has negative temperature coefficient constantly and it was found to be consistently increased with respect to temperature. This elevated view of the Gibbs free energy showed the flat character of the continual chemical reaction of compound.
| (K) |  |  |  | Gibbs free energy  KJmol‑1 |
| 100.00 | 282.07 | 68.38 | 4.87 | -6833.13 |
| 200.00 | 340.96 | 106.12 | 13.58 | -21210.4 |
| 298.15 | 390.68 | 145.80 | 25.93 | -43444.3 |
| 300.00 | 391.58 | 146.56 | 26.20 | -43941.8 |
| 400.00 | 439.16 | 185.35 | 42.83 | -74097.2 |
| 500.00 | 484.19 | 218.40 | 63.07 | -109137 |
| 600.00 | 526.47 | 245.23 | 86.30 | -147052 |
| 700.00 | 565.96 | 266.89 | 111.95 | -186711 |
| 800.00 | 602.79 | 284.61 | 139.55 | -227548 |
| 900.00 | 637.19 | 299.33 | 168.77 | -269228 |
| 1000.00 | 669.38 | 311.74 | 199.34 | -311541 |
Table 10: Specific heat capacity, entropy and enthalpy at different temperatures for Isonicotinohydrazide
4.11. Molecular Docking Study
Anthranilate phosphoribosyltransferase (AnPRT) belongs to phosphoribosyltransferase (PRT) enzyme family and is an attractive enzyme in the field of drug discovery and tuberculosis therapy, since it is essential for the biosynthesis of tryptophan in Mycobacterium tuberculosis (Mtb) [48]. Each chains of AnPRT consist of a smaller N-terminal domain and a larger C-terminal domain. C-terminal domain contains a central seven-stranded β-sheet surrounded by eight α-helices [49] and N-terminal domain consists of six α-helices. In each chain, two anthranilate binding sites were present, one of which is located in the hinge region between the N-binding site and an another site situated farther to the phosphoribosyl group of 5ʹ-phosphoribosyl-1ʹ-pyrophosphate (PRPP). Inhibitors like anthranilate and flouroanthranilate have been observed to bind to Mtb-AnPRT in the abovementioned active sites, located at the entrance to the anthranilate tunnel [50]. In order to understand how the title molecule inhibits and hence increases the catalytic efficiency of Mtb-AnPRT enzyme, a docking study was performed to fit the title compound into the active centre of the Mtb-AnPRT enzyme. High resolution crystal structure of Mtb-AnPRT (PDB ID: 1ZVW) [51] was used as the receptor enzyme and AutoDock4 software was used to perform all molecular docking simulations. For the calculations, water molecules were removed and polar hydrogen atoms were added to the receptor molecule. The active centre around the N-binding site of the receptor was defined by incorporating all amino acid residues within a radius of sphere 7 Å centered on the co-crystallized inhibitor (PRPP).
In order to model the interaction pattern between receptor and the ligand, Lamarckian Genetic Algorithm (LGA) available in AutoDock was employed. Out of ten docked confirmations obtained, one which has lowest binding energy (-7.26 kcal/mol) was selected and analyzed for detailed interactions using Discover Studio Visualizer4.0 software and Pymol. The predicted binding mode for the title molecule showing significant inhibition and the amino acid residues which form hydrogen bonds with the ligand were labeled in Figure 11. The title compound shows hydrogen bond formation with residues Asp111, Thr115 and Ser143. The electrostatic contacts are driven by Mg2+ ions present in the active site. The amino group in the title molecule acts as hydrogen donor and forms hydrogen bond with Asp111, while it acts as hydrogen acceptor in making hydrogen bond with Thr115. The docked binding pose of the title molecule also speculates the formation of another strong hydrogen bond with residue Ser143 where carbonyl oxygen is acting as hydrogen acceptor. Surface representation of the receptor with the ligand bound is shown in Figure12 from which it can be seen that, the ligand is bound well inside the active pocket of the enzyme. Estimated free energy of binding shows that the title compound has good affinity towards the Mtb-AnPRT enzyme. The results from the molecular docking study revealed that the title molecule may inhibit catalytic efficiency of Mtb-AnPRT enzyme.
In order to study the chemical as well as pharmaceutical property of the compound, the molecular spectroscopic analysis has been carried out. Here, all the investigations made on the molecule proved the root cause of the antibiotic property. The vibrational analysis emphasized activeness of the compositional parts of molecule which also confirmed the position and presence of all bonds of compound. The frontier molecular orbital interaction profile was found to be favored for the energy distribution over different entity of the molecule. The UV-Visible analysis stressed the chromophores activity from electronic excitation absorption for the functional groups. The present compound was identified as pharmaceutically active and background causes committed for the inducement of antibiotic property was determined. The inhibition catalytic efficiency of the title molecule was tested from molecular docking studies.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
