
Case Report | DOI: https://doi.org/10.31579/2641-0419/151Copyright
*Corresponding Author: Zed Seedat, Florida Atlantic University College of Medicine Internal Medicine Residency Program 777 Glades Road, BC-71 Rm 335 Boca Raton, Florida, 33431
Citation: Zed Seedat., Gopika Dasari., Seth Baum., (2021) Sitosterolemia: A Rare Case with Broad Implications. J. Clinical Cardiology and Cardiovascular Interventions, 4(8); Doi:10.31579/2641-0419/151
Copyright: © 2021 Zed Seedat, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 09 March 2021 | Accepted: 09 April 2021 | Published: 16 April 2021
Keywords: sitosterolemia; genetics; cholesterol; coronary artery disease; low-density lipoprotein cholesterol
Sitosterolemia is an ultra-rare autosomal recessive dyslipidemia characterized by mutations in genes encoding the ATP-binding cassette (ABC) G5/8 transporters. We describe the case of a 20-month-old female presenting with xanthomas and serum low density lipoprotein cholesterol of 657 mg/dL. Diagnostic workup revealed a previously undescribed sitosterolemia-causing mutation. After elimination of dietary sterols and initiation of ezetimibe therapy, the patient’s xanthomas resolved, and serum low density lipoprotein cholesterol was reduced to 104 mg/dL. Importantly, pathologically elevated serum phytosterols were found in each of the proband’s heterozygous parents. Elevated phytosterols, an established cause of atherosclerosis, are typically unrevealed by standard lipid testing. As heterozygous mutations for ABCG5/8 are relatively common, this has implications for a broader population than the ultra-rare sitosterolemia cohort. Thus, insights gleaned from this case highlight underappreciated matters in the prevention of atherosclerotic disease in both heterozygous and homozygous carriers alike.
Abbreviations
ABC : ATP-binding cassette
ALT : Alanine transaminase
AST : Aspartate transaminase
ASCVD : Atherosclerotic cardiovascular disease
GC/MS : column gas liquid chromatography/mass spectrometry
HDL-C : high density lipoprotein cholesterol
LDL-C : low density lipoprotein cholesterol
NPC1L1 : Niemann-Pick C1-Like 1
TG : Triglycerides
A 20-month-old girl presented to our preventive cardiology/lipidology clinic following an 8-month history of xanthomatous lesions on her buttocks and in her popliteal creases (figures 1-2). Both pediatrics and dermatology had identified the lesions as xanthomas and accordingly ordered a lipid panel. The patient’s cholesterol levels were severely elevated, leading her clinicians to seek our input. The patient’s history was significant for normal growth and development and the absence of any observed exertional symptoms. Family history was remarkable for the absence of premature vascular disease. Physical examination revealed xanthomas in popliteal creases, and on the ankles and buttocks; corneal arcus and xanthelasmas were absent. Cardiac, pulmonary, and rheumatologic examinations were normal. Splenomegaly was absent.
Homozygous and heterozygous familial hypercholesterolemia, sitosterolemia, and cerebrotendinous xanthomatosis comprise the differential diagnosis for pediatric patients presenting with xanthomatous disease [1]. Neurologic findings were absent; thus, cerebrotendinous xanthomatosis was highly unlikely. As familial hypercholesterolemia is nearly always autosomal dominant, while Sitosterolemia is always autosomal recessive, the absence of a family history of ASCVD significantly favors the diagnosis of sitosterolemia.
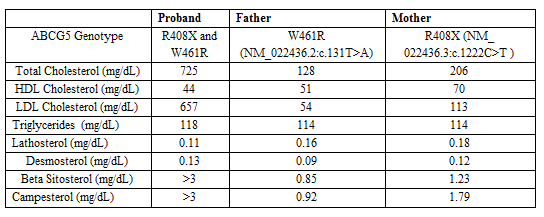
A lipid panel following the identification of xanthomas revealed: total cholesterol 725 mg/dL, low density lipoprotein cholesterol (LDL-C) 657 mg/dL, high density lipoprotein cholesterol (HDL-C) 44 mg/dL, and triglyceride (TG) 118 mg/dL. Complete blood count revealed microcytic anemia. Reticulocyte count, lactate dehydrogenase, AST, ALT, total bilirubin and haptoglobin were within normal limits. Carotid artery duplex ultrasound and echocardiogram were unremarkable.
Targeted lipid gene sequencing and column gas liquid chromatography/mass spectrometry (GC/MS) testing for serum sterol concentrations were obtained on the patient and both parents. Compound heterozygous coding mutations in ABCG5 (R408X and W461R mutations) were identified in the proband. The previously undescribed W461R mutation was also identified in the patient’s father, while the R408X mutation was present in the mother. The patient’s phytosterol levels were markedly elevated with beta-sitosterol and campesterol above the detectable limit of > 3 mg/dL (normal, < 0.8 mg/dL and < 1.03 mg/dL respectively); lathosterol and desmosterol were normal at 0.11 mg/dL (normal, < 0.81 mg/dL) and 0.13 mg/dL (normal, < 0.35 mg/dL) respectively. Significant elevations in serum beta-sitosterol and/or campesterol levels were found in both parents. It is important to note that LDL-C was not significantly elevated in either parent (Table 1).
Prior to genetic assessment, the patient had initially been given a provisional diagnosis of homozygous familial hypercholesterolemia. Pending evaluation at our clinic, she was advised to make dietary changes including eliminating pizza, cheese, and meat [2]. Six weeks later, the patient’s LDL-C level decreased approximately 50% to 381 mg/dL.
After definitively (genetically) diagnosing sitosterolemia, we advised the patient’s parents to further modify her diet by eliminating all phytosterol-rich food. Ezetimibe 5mg daily was also initiated [2]. Given the absence of exertional symptoms, further imaging with CT coronary angiography was not performed.
Several weeks after the patient had made stringent dietary changes and started ezetimibe 5 mg daily, a repeat lipid panel revealed marked reductions in total and LDL cholesterol, 153 mg/dL and 104 mg/dL respectively. Additionally, the patient’s cutaneous xanthomas resolved. Because of socio-economic challenges and her parent’s philosophical reservations regarding prescription medications, the patient’s ezetimibe therapy was stopped, and she has not yet returned to the clinic. The patient’s family has been repeatedly contacted and strongly encouraged to resume therapy. Her parents have also been counseled to undergo their own cardiovascular consultations for lipid management and risk reduction.
Sitosterolemia is an ultra-rare autosomal recessive lipid disorder caused by mutations in the ATP Binding Cassette (ABC) transporters G5/8 [3]. Present in small intestine enterocytes and biliary tract epithelial cells, these ABC transporters participate in the efflux of both cholesterol and the biochemically similar dietary phytosterols, including sitosterol and campesterol (figure1,2and 3) [3].

Figure 1: Xanthomatous lesions over right ankle. Classic appearing pediatric xanthomas are seen.
A new sitosterolemia-causing mutation (NM_022436.2:c.131T>A) was discovered in this case. Additionally, significant elevations in serum phytosterols were identified in both heterozygote parents. This finding highlights an unrecognized increased risk of atherosclerotic disease in heterozygous carriers of sitosterolemia-causing mutations, and the need to identify and treat these not-so-rare individuals.
Disclaimers: The words expressed in the submitted article are original and have not been influenced by any institution or funding.
Source(s) of Support: No sources of support were used for the submitted article.
Disclosure: Drs. Seedat and Dasari have nothing to disclose and no financial conflicts of interest. Dr. Baum has consulted/served on advisory boards for Amgen, Madrigal, Sanofi, Merck, Allergan, and Esperion.
Acknowledgements - We thank Dr. Ernst J. Schaefer and Boston Heart Diagnostics for analysis of serum phytosterols. We thank Johanna Lora for technical assistance. We thank Vivian Bomie Lee and BomieArt for illustrative assistance.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
