
Research Article | DOI: https://doi.org/10.31579/2768-0487/161
Department of Cytogenetics, Gujarat Cancer and Research Institute, Ahmedabad, Gujarat, India.
*Corresponding Author: Pina J. Trivedi, Department of Cytogenetics, Gujarat Cancer and Research Institute, Ahmedabad, Gujarat, India.
Citation: Pina J. Trivedi, Patel D, (2025), Simultaneous Occurrence of Additional Chromosomal Abnormalities in a Case of Acute Promyelocytic Leukaemia Resulting in Rapidly Fatal Disease, Journal of Clinical and Laboratory Research, 8(1); DOI:10.31579/2768-0487/161
Copyright: © 2025, Pina J. Trivedi. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 01 November 2024 | Accepted: 02 December 2024 | Published: 01 January 2025
Keywords: disease; red blood cell; genetic; cancer
Reciprocal translocation t(15;17)(q22;q21) is a hall mark of Acute Promyelocytic Leukemia (APL). Approx. 23–43 % of cases show additional chromosomal abnormalities. Here we report a case of a 33-year-old woman with APL with the typical t(15;17) translocation, and with a secondary changes deletion 7q as an additional abnormality. Our patient presented with characteristic clinical and morphological features of APL. Conventional cytogenetics and Fluorescence In Situ Hybridization (FISH) using dual color dual fusion probe analysis confirmed the t(15;17)(q22;q21). Patient did not achieve remission and was expired within three days of diagnosis. For conventional cytogenetic study, short term culture of bone marrow and Giemsa Trypsin Giemsa banding were carried out according to standard protocols. Cytogenetic nomenclature was carried out using ISCN 2020 guidelines. Our patient presented with a rare cytogenetic profile and rapidly progressive disease. According to our extensive literature search, this was the first case of APL having pathognomonic t(15;17) along with monosomy 6, deletion 7q.
Acute Promyelocytic Leukemia (APL) is a subtype of AML, characterized by fusion gene PML-RARa and this fusion protein acts as transcriptional repressor inhibiting normal myeloid differentiation, cure rates with targeted therapy is high [Sunil Girish Iyer 1 2023, Shahid M et al 2016]. Additional Chromosome Aberrations(ACA) occur in 29–43% APL patients at diagnosis along with translocation 15 and 17 [Sanz MA et al 2014 & Liu Y et al, Ma L, et al. 2020]. There are debatable results for prognostic impact of ACA in APL [Pantic M et al, de Botton S et al 2000]. ACA are randomly encountered either at diagnosis or during therapy [Haidary AM, et al 2022]. Trisomy 8 is the most common numerical abnormality observed in 33% to 53% of secondary changes. Patients show ACA with low platelet counts, intermediate and high-risk disease, and the presence of coagulopathy are the main characteristics & clinicopathological feature, which is also observed in present study [José Cervera,1 et al 2006]. Study showed that secondary chromosomal abnormalities are less specific and occur in an addition to the primary abnormalities. This means that any APL cases confirmed by the detection of t(15;17)(q22;q12) with myelodysplastic changes should be diagnosed as belonging in the AML category. A comprehensive study results showed that in primary MDS, the translocation of chromosomes 15 and 17 was not noted among the 31 AML transformed from MDS (MDS/AML) [de Souza Fernandez T et al 2000]. So it is a question of debate that suppose an individual is diagnosed as having APL with MDS, which condition developed first, the APL or the MDS. Myelodysplastic episodes that precede the onset of APL are linked to a poor prognosis. Clinicians would apply the standard treatment for APL, including All-Trans Retinoic Acid (ATRA) and Arsenic Trioxide (ATO), for better outcomes, if additional chromosomal abnormalities observed in an APL case during the disease progression. Thus, because the diagnosis is critical to the treatment plan, it would be prudent to determine whether the APL has evolved from MDS (MDS/APL) and whether the APL occurred with additional chromosomal abnormalities [Osamu Imataki* and Makiko Uemura 2016]. AML and MDS showed the most common cytogenetic abnormalities is of chromosome 7, either monosomy (−7) or deletion of the long arm (del(7q). This occurs in ~ 8% of de novo AML and ~10% of de novo MDS. In International Prognostic Scoring System, Monosomy 7 is classified as poor prognostic criteria as relapse rate at one year is 82%, and the 7-yr actuarial event-free survival is 6%; after an allogeneic bone marrow transplantation, -7 is predictive of an unfavourable outcome [François Desangles et al 1999]. Rarely, cases have been reported with clinical and morphological features of APL where the cytogenetic evaluation revealed presence of t(15;17) along with other recurrent genetic abnormalities. Here in present study we observed a female with deletion 7q along with t(15;17) and monosomy of 6.
Case Details:
We report a patient with APL who had translocation 15 and 17 along with deletion of 7q prior to the course of therapy. A 33-years-old female suffering from low grade fever, weakness was referred to Gujarat Cancer and Research Institute, Ahmadabad, India. The laboratory investigations revealed White Blood Cell count 75.11 X103/cmm, Red Blood Cell count 1.46 X 106 Cells/µL, Hemoglobin- 4 gm/dl, and Platelets 5 x 103/cmm. PS RBCs are predominantly normocytic normochromic. Retic-0.51%. WBCs series show leukocytosis with increased blast & promyelocytes. Blast+Promyelocytes-91, P-01, L-05, M-03. Platelets reduced. MP not seen. Bone Marrow aspiration report showed Hypercellular marrow with altered M:E Ratio, Promyelocytes-92% Myelocytes 5 %, Late Normoblasts-3 %, no megakaryocytes, erythropoiesis suppressed, PAS Stain Negative, Sudan Black B-Positive, Blasts are medium to large sized have high N:C ratio, fine chromatin and moderate cytoplasm with numerous azurophilic granules. Occasional faggot's cells seen. Erythroid precursors (3%) suppressed.
Immunophenotyping:
A result of Immunophenotyping of bone marrow was 95% blasts gated using CD45 PerCP vs. side scatter. The blasts mainly expressed myeloid markers MPO, CD13, CD33, CD117 and CD15 along with CD34. Co expression of CD7 was seen. Final diagnosis was Acute Promyelocytic Leukemia (APML).
Other Investigation:
CT SCAN of Brain:
Presence of approx. 27x26x20 mm sized hyper dense lesion (avg CTHU +61) is noted involving basal ganglia region. Presence of few similar characteristic region is noted adjacent to it. Presence of perilesional hypo density is noted. It causes mass effect in form of compression of frontal horn of left lateral ventricle. Presence of ill-defined hyper dense area (avg CTHU + 52) is noted involving right cerebellar hemisphere. P/o intraparenchymal bleed appears likely. Basal cisterns appear normal. No e/o subarachnoid hemorrhage seen. Bony skull vault appears normal. Findings suggested that Hyper dense lesion involving basal ganglia region as described above.
Conventional Cytogenetics:
For conventional cytogenetic study bone marrow sample was collected in sterile Sodium Heparinized vaccuate & short term culture was carried out as per standard protocol. Slides were banded using Giemsa Trypsin G banding technique. Good morphology metaphases were captured in Zeiss Automatic Karyotyping system(Metafer) and analysis using IKAROS software and karyotype description was done using ISCN 2020 guidelines.
Fluorescence in situ hybridization (FISH):
FISH was performed using PML-RARa LSI probe. In LSI PML-RARa probe, RARa gene was tagged with SG and PML gene tagged with SO. Epi-fluorescence microscope (AXIO Imager.Z2, Zeiss, USA) equipped with appropriate filter sets was used for FISH study. Image capturing and processing were carried out using an ISIS FISH imaging system (MetaSystems, Germany). Further D7S522 (7q31, spectrum red)/ CEP7 (spectrum green) probes (Abbott Molecular). Two hundred nuclei were counted and the percentage of cells with del(7q) was calculated. The positive cutoff value for del(7q) established in our laboratory is 3.6%.
Conventional Cytogenetic:
Conventional chromosome analyses at diagnosis of GTG banded metaphase were carried out. Total 20 metaphases were karyotyped. All metaphases showed 45,XX,-6, del(7q),t(15;17)(q22;q21), [20]. Partial karyotype of t(15;17), deletion 7q and monosomy 6 is shown in (Figure.1).

Figure 1: G banded Partial karyotype of t(15;17), deletion 7q and monosomy 6.
FISH:
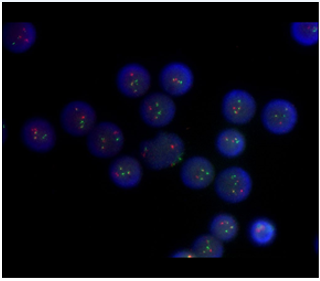
Figure 2: FISH results with PML-RARa probes results revealed 1O1G2F signal pattern in interphase cells which showed that there was translocation of PML and RARa gene.
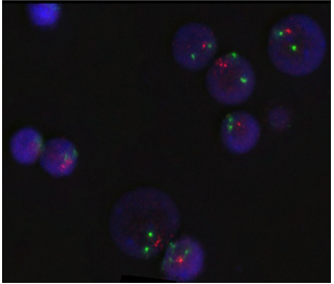
Figure 3: FISH results with deletion 7q probes showed loss of q arm of chromosome 7 as (SG
Cytogenetic study results are diagnostically important for confirmation of diagnosis and choice of treatment in APL [Miguel A. Sanz, et al 2019]. It is the most powerful single prognostic factor for outcome in AML and the most useful guide available for stratification and planning post remission treatment in this disease [ José Cervera, et al 2007]. A sensitive techniques Fluorescence in Situ Hybridization (FISH) helps in differential diagnosis or therapeutic planning and provides rapid results. Combining FISH and cytogenetic results enables a more precise evaluation of disease prognosis, diagnosis, classification, risk stratification, and patient treatment [Hemani Jain1 et al 2023] Trisomy 8 is the most common numerical chromosomal abnormalities generally observed as ACA in APL that can be either as a sole or in combination with other abnormalities [Baccarani M et al 2017, Safaei A, et al 2018]. In present study we observed monosomy of chromosome 6 which is also a very rare abnormality with t(15;17). Generally, chromosome 6 is reported as either trisomy 6 or deletion 6q in majority of leukemia cases. Structural chromosomal abnormalities include 9q, 7q, 1p, 11q, 3q, 20q, 17p and complex three‐way variants translocations involving chromosome 15, 17 and another chromosome with resultant PML‐RARA fusion [Safaei A et al 2018]. Complete or partial deletions of chromosome 7 (-7/del7q) belong to the most frequent chromosomal abnormalities in myeloid neoplasm (MN) and are associated with a poor prognosis. There is no any clear relation with -7/del7q and the genes responsible for the leukemogenic properties. Haploinsufficient (HI) genes contained in the deleted regions of chromosome that may create clonal vulnerabilities. Therefore, HI genes are potential targets of synthetic lethal strategies. Despite decades of studies, the various candidate genes responsible for the leukemogenic properties of -7/del 7q have been proposed, but their role has not been completely elucidated, and no targeted therapies have been conceptualized [Minako Mori et al 2023]. The mechanisms by which -7/del7q may induce leukemic transformation are multiple and there are not any compatible hypotheses for leukemogenesis. These concepts implicate loss of heterozygosity (LOH), haploinsufficiency (HI) of tumor-suppressor genes (TSGs) and somatic rescue of suppressive germline variants located within the deleted regions [O’Neil NJ et al 2017 & Pan R et al. 2017]. ATRA lead the patient into molecular remission. Present results propose that an assessment of ACA in APL would contribute to the clinical decisions regarding whether to treat disease with ATRA and cytotoxic agents. It would be of interest to know the extent of cytogenetic abnormality in the patients regarding to mixed leukemia. One or more additional cytogenetic abnormalities other than PML-RARA could account for the biological malignant grade and prognostic index [Osamu Imatak et al 2016]. There is no known science which can change the genetic machinery of the cancerous cells is that it may provoke uncontrolled increase in proliferating immature cells or there is arrested maturation. [Horibata S et al, 2020]. Chromosomal analysis using conventional karyotypic by Giemsa banding revealed presence of monosomy 6, deletion 7q along with the pathognomonic t(15;17). Which proved that patient had rapidly progressive and fatal disease, having typical clinical and morphological features of APL. Such karyotypic profile has never been reported elsewhere in the literature.
So present study concludes that though there is identification of targeted therapy like ATRA and ASO in APL cases, the high risk of early death without prompt initiation of treatment at first clinical suspicion, and dedicate a special focus to novel agents and future directions to improve cure rates and quality of life in patients affected by additional chromosomal abnormalities along with APL.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
