
Case Report | DOI: https://doi.org/10.31579/2692-9392/175
1 David Geffen School of Medicine at UCLA, Los Angeles, CA.
2 Department of Medicine, Cedars-Sinai Medical Center, Los Angeles, CA.
3 Department of Medicine, Olive View Medical Center, Sylmar, CA.
*Corresponding Author: Rintu Saju, B.S. David Geffen School of Medicine at UCLA, Los Angeles, CA.
Citation: Rintu Saju, Natalie Bransky, Stanley Yuan and Nasser Mikhail (2023), Sheehan Syndrome Presenting with Life Threatening Hyponatremia, Archives of Medical Case Reports and Case Study, 7(3); DOI:10.31579/2692-9392/175
Copyright: © 2023, Rintu Saju. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 13 June 2023 | Accepted: 20 June 2023 | Published: 27 June 2023
Keywords: sheehan syndrome; pituitary necrosis; hyponatremia; hyperlipidemia
A 58-year-old gravida 4 para 4 woman with a history of hyperlipidemia was sent to the emergency room by her primary care physician after routine labs showed hyponatremia to 119 mmol/L. Patient was asymptomatic, reporting no headache, dizziness, confusion, visual disturbances, numbness, weakness, chest pain, dyspnea, palpitation, vomiting, diarrhea, or abdominal pain. She reported daily fluid intake of 1-1.5L. She reported a history of uncontrolled hyperlipidemia (reportedly to the 900s) and was previously trialed on several different statins but discontinued due to myopathy. She was currently taking fish oil supplementation for her hyperlipidemia. She was not taking any other medications
A 58-year-old gravida 4 para 4 woman with a history of hyperlipidemia was sent to the emergency room by her primary care physician after routine labs showed hyponatremia to 119 mmol/L. Patient was asymptomatic, reporting no headache, dizziness, confusion, visual disturbances, numbness, weakness, chest pain, dyspnea, palpitation, vomiting, diarrhea, or abdominal pain. She reported daily fluid intake of 1-1.5L. She reported a history of uncontrolled hyperlipidemia (reportedly to the 900s) and was previously trialed on several different statins but discontinued due to myopathy. She was currently taking fish oil supplementation for her hyperlipidemia. She was not taking any other medications.
Patient was bradycardic to 48 beats per minute on presentation with all other vital signs normal (blood pressure: 122/65, respiratory rate: 12, and temperature: 36.4°C). She weighed 53kg and had a body mass index of 22.64 kg/m2. On clinical examination, patient was well appearing, alert and oriented, without neurologic deficits, and without any other notable findings. She appeared euvolemic on exam.
Complete blood count was revealing for mild leukopenia at 3.8 K/cumm (normal range: 4.5-10 K/cumm), normocytic anemia with hemoglobin of 10 g/dL (normal range: 12-14.6 g/dL), hematocrit of 28.2% (normal range: 36-44%), mean corpuscular volume of 85.1 fL (normal range: 82-97 fL), and a normal platelet count of 280 K/cumm (normal range: 180-382 K/cumm). Metabolic panel showed severe hyponatremia at 119 mmol/L (normal range: 136-144 mmol/L) and hypochloremia to 87 mmol/L (normal range: 97-108 mmol/L) with normal kidney and liver function. Thyroid stimulating hormone (TSH) was normal at 1.415 uIU/mL (normal range: 0.350-4.940 uIU/mL). Lipid panel showed elevated cholesterol of 767 mg/dL (normal range: 125-199 mg/dL) and low-density lipoprotein at 693 mg/dL (normal range: <99>
Patient was admitted to the intensive care unit (ICU) and was started on fluid restriction and sodium chloride tablets 2 grams three times daily for treatment of hyponatremia. Repeat serum sodium at this time was 117 mmol/L (normal range: 136-144 mmol/L) and was unchanged after supracentrifugation. She remained bradycardic in the ICU, reaching a nadir of 43, and was intermittently hypotensive, with lowest blood pressure to 81/52.
Given the presence of this new onset hypotension coupled with euvolemic hyponatremia, adrenal insufficiency was considered, and subsequent workup revealed low morning serum cortisol at 2.5 mcg/dL (normal range: 3.7-19.4 mcg/dL). Cosyntropin stimulation test showed a rise in cortisol from 3.7 mcg/dL (index time) to 10.1 mcg/dL (60 minutes) confirming diagnosis of adrenal insufficiency since peak serum cortisol after cosyntropin stimulation did not reach the cutoff of 18 mcg/dL [1]. Free thyroxine (fT4) and triiodothyronine (T3) levels checked at this time were undetectable with a normal TSH level (normal range: 1.554 uIU/mL). She was started on hydrocortisone (20 mg in the morning and 10 mg in the evening) and levothyroxine (88 mcg once daily). Urine output subsequently increased, urine sodium decreased (15 mmol/L), and serum sodium levels improved to 129 mmol/L (normal range: 136-144 mmol/L). Salt tablets were discontinued, and patient received spot doses of vasopressin to avoid rapid sodium correction. Pituitary panel showed low insulin-like growth factor-2 of 204 ng/mL (normal range: 267-616 ng/mL) and prolactin of 1.8 ng/mL (normal range: 4.8-23.3 ng/mL), and normal adrenocorticotropin hormone, follicle-stimulating hormone, and luteinizing hormone (Table 1). MRI brain revealed a normal size pituitary fossa that was predominately filled with cerebrospinal fluid with minimal residual enhancing pituitary tissues mainly in the postero-inferior aspect of the sella (Figure 1) [2].

Table 1: Selected Laboratory Values

Figure 1: Appearance of an empty sella on gadolinium enhanced T1-weighted magnetic resonance images in A) coronal and B) sagittal view. Images were obtained from Kaplun et. al.2
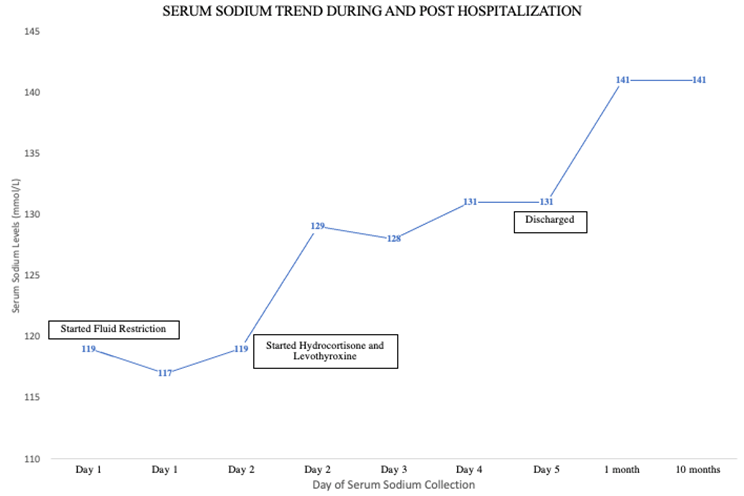
Figure 2: Serum Sodium Levels During and Post-Hospitalization
Upon further questioning, patient reported a history of massive bleeds during a forceps-assisted vaginal delivery 28 years prior. She reported being amenorrheic since that time. Given this history and constellation of findings suggestive of hypopituitarism, a diagnosis of Sheehan syndrome was made. The patient was discharged on hydrocortisone (15 mg every morning and 10 mg every evening) and levothyroxine (88 mcg daily). Patient remained well at the 10-month follow-up, with her anemia resolved, LDL levels decreased to 290 mg/dL (normal range: <99>
We report a case of Sheehan syndrome in a patient presenting with asymptomatic hyponatremia. Patient had a history of obstetric complication resulting in postpartum hemorrhage, subsequent amenorrhea, and clinical, laboratory, and MRI findings consistent with hypopituitarism.
Sheehan syndrome is a rare, parturition-related complication resulting in acute pituitary infarction and hypopituitarism [3]. The condition was named after the works of Harold L. Sheehan, who in 1937 showed that ischemia, and not puerperal sepsis or mycotic bacterial emboli as was previously described [4,5], leads to the acute glandular necrosis [6]. In a 1996 estimation by the World Health Organization, Sheehan syndrome affected roughly three million women worldwide, with a vast majority of cases likely concentrated in developing countries where access to adequate obstetrics care is more limited [7-9]. In comparison, developed countries have lower, but a non-negligible rate of Sheehan syndrome estimated to be around 5.1 cases per 100,000 population [10]. The possibility of diagnostic delay and potential misdiagnosis may make this an underrepresentation of the actual disease prevalence [11].
Sheehan’s work described that peripartum bleeding is an important risk factor for pituitary infarction [6]. While postpartum hemorrhage remains the most common etiology, rare cases of non-obstetric sentinel bleeding events in a pregnant person have also been described [12-14]. The pituitary gland undergoes significant changes during pregnancy, including estrogen-mediated lactotroph hypertrophy, resulting in a gland almost 120-136% of its normal size by the end of pregnancy [15-17]. The growing pituitary requires a robust vasculature that is unfortunately also highly susceptible to ischemia and infarction, even with minute alterations to the pituitary intravascular pressure [3,18]. The pituitary receives its blood supply primarily through the inferior and superior hypophyseal arteries. The inferior hypophyseal artery feeds much of the posterior pituitary, while the superior treads through the hypothalamus and forms the low-pressure portal capillary circuit that eventually feeds the anterior lobe [19,20]. The growing anterior lobe can cause physical compression of the superior hypophyseal artery, which in combination with the smaller sella turcica that is reported among some Sheehan syndrome patients, can render the pituitary vulnerable to infarction in the setting of an acute bleed [8,21].
More so than the amount of bleeding itself, severity and duration of shock may be a stronger predictor of pituitary infarction.22 However, the exact mechanism of how infarction develops remains controversial to date. Studies have shown potential roles of vascular compression, cytokine-mediated vasospasm in the setting of hypovolemic shock, primary thrombosis in the setting of hypercoagulation or dissemination intravascular coagulation, and certain genetic factors as possible mediators of glandular necrosis [23-25]. Additionally, acute necrosis may also release tissue content which in turn cause immune-mediated destruction of the gland with time [26]. Progressive decline of pituitary function has been documented among Sheehan patients, as is the presence of anti-pituitary antibodies in upwards of 60% of cases that persisted long after initial insult [26]. With time, the necrotic debris is replaced by cerebrospinal fluid, giving it the characteristic appearance of an empty sella on radiographic evaluation [27].
Destruction of more than 70% of the gland results in partial or panhypopituitarism, with the latter being the most common among patients [28,29]. Although both the anterior and posterior glands are affected, posterior gland dysfunction causing diabetes insipidus is rarely described [24,30]. Rather, a varying degree of anterior pituitary dysfunction typically occurs, resulting in a wide array of clinical presentations, from isolated hormonal deficiencies to signs of panhypopituitarism [24]. Somatotrophs and lactotrophs are the most susceptible to ischemic damage due to their anatomic location within the pituitary and are relatively lost in a vast majority of patients with Sheehan syndrome, while patients will display varying degrees of gonadotropic, thyrotropic, and corticotropic deficiencies [24]. Additionally, patients may be asymptomatic for a long time or have non-specific symptoms (e.g., fatigue, weakness, or anorexia) that prolong diagnosis until an acute stressor (e.g., infection, trauma, or surgery) leads to adrenal crisis or myxedema coma [3,21,24]. All in all, the heterogeneity of presentation makes timely diagnosis challenging, delaying it between 2 and 40 years post-delivery [31].
In our patient, diagnosis was delayed by roughly 30 years postpartum. Postpartum hemorrhage and presence of amenorrhea were important pieces of history that helped clue into the diagnosis. Amenorrhea along with agalactia postpartum are characteristic of Sheehan syndrome, although central gonadotropic levels may be normal, as with our patient [21,32]. Similarly, our patient had normal TSH levels, which may be paradoxically normal or increased in Sheehan patients due to low thyroid hormone levels [24]. However, circulating TSH undergoes sialylation which may decrease clearance and its metabolic activity [24]. It should be emphasized that diagnosis of central hypothyroidism may be missed in institutions where it is standard to only obtain fT4 results if there is an abnormal TSH. Indeed, in our case, diagnosis of pituitary hypothyroidism was delayed almost 24 hours because we relied only on the normal TSH without checking free T4 levels. fT4 should be sought after in the correct clinical context, especially if there is high index of suspicion for thyroid abnormalities, even in cases where TSH is normal.
Hyponatremia is a common electrolyte abnormality noted among patients with Sheehan syndrome and is seen in up to 33-69% of cases [33]. Multiple mechanisms have been implicated, including increased effects of antidiuretic hormone secondary to glucocorticoid deficiencies and hypotension, and hypothyroidism-induced decrease in free-water excretion through mechanisms irrespective of vasopressin’s effects [24,33]. Thyroid and steroid hormone replacement was cornerstone in helping normalize our patient’s serum sodium levels. Normocytic anemia, as in our patient, is another common complication of Sheehan syndrome [24]. Cortisol and thyroid hormone are pertinent in erythropoietin synthesis and maintaining its biological efficacy. Replacement with steroid hormone and thyroxine is critical in reversing this hematologic complication [24].
Our patient also had a long-standing history of poorly controlled hyperlipidemia. It is unclear whether the patient’s hyperlipidemia is a primary process or an acquired complication of Sheehan syndrome. Several endocrinopathies associated with Sheehan syndrome, including growth hormone deficiency and hypothyroidism, can contribute to the increased levels of obesity and dyslipidemia seen in this population [34]. Growth hormone normally inhibits the conversion of inactive cortisone to active cortisol by 11  -hydroxysteroid dehydrogenase type I. Deficiencies can increase formation of active cortisol, contributing to weight gain and dyslipidemia [34]. In one case report, thyroid hormone replacement led to correction of a patient’s hyperlipidemia, although other reports suggest that growth hormone replacement, and not thyroxine alone, may be crucial for correcting lipid derangements [35,36].
-hydroxysteroid dehydrogenase type I. Deficiencies can increase formation of active cortisol, contributing to weight gain and dyslipidemia [34]. In one case report, thyroid hormone replacement led to correction of a patient’s hyperlipidemia, although other reports suggest that growth hormone replacement, and not thyroxine alone, may be crucial for correcting lipid derangements [35,36].
Standard treatment protocol recommends starting glucocorticoid first before initiating thyroid replacement to avert a potential adrenal crisis [19]. Hydrocortisone is most often used, and should be administered twice a day, with the higher dose taken in the morning to mimic the natural diurnal pattern. Thyroxine replacement is also recommended using levothyroxine, with lower doses recommended in older individuals and in those with evidence of coronary artery disease. Although growth hormone may improve the metabolic profile in patients with Sheehan syndrome, no formal guidelines exist regarding its replacement, and the decision should be made based on risks, benefits, and cost of treatment. Sex hormone replacement can be considered in premenopausal women due to increased risk for osteoporosis [19].
Ultimately, a diagnosis of Sheehan syndrome is made based on clinical history, which includes history of significant postpartum hemorrhage, lactation failure, and amenorrhea; an endocrine panel suggestive of isolated or panhypopituitarism; and related radiographic findings of either partial or completely empty sella [21]. Our patient met several of these criteria, including having a history of postpartum hemorrhage, amenorrhea, anterior pituitary dysfunction with associated sequalae, and evidence of an almost completely empty sella on MRI. Steroid and thyroid hormone replacement therapy were crucial components of her treatment.
Patients with Sheehan Syndrome have 1.2 to 2.7 times the mortality rate compared to the general population [37]. Potential delay in diagnosis significantly impacts the development of adverse metabolic conditions and increases risk for adrenal crisis and myxedema coma [24]. As Sheehan syndrome is rare in the developed world, clinicians may be less familiar with the subtle clinical features associated with this syndrome. Patient’s presentations are rarely overt and are heterogenous in nature, making diagnosis challenging. Taking a thorough history, including a detailed peripartum history, and considering Sheehan syndrome in the differential diagnosis for patients presenting with hyponatremia and long-standing hyperlipidemia may be helpful in abridging the time between initial insult and diagnosis. Timely hormone replacement can improve quality of life and provide overall mortality benefits for affected patients.
We thank the patient for giving us consent to publish this case and disseminate additional literature on the wide presentation range of Sheehan syndrome. We also thank everyone directly and indirectly involved in the patient’s care, and in particular, the primary medicine team, the intensive care unit team, and the consulting endocrinology and nephrology team for all their hard work and diligence throughout this case in service to the patient.
Contributors
All authors made individual contributions to authorship.
All authors reviewed and approved the final manuscript.
Funding
No public or commercial funding
Disclosure
None declared
Informed Patient Consent for Publication Statement
Data Availability
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
