
Case Report | DOI: https://doi.org/10.31579/2768-0487/103
Department of Laboratory Medicine, Korea University Medical Center Guro Hospital, Korea.
*Corresponding Author: Jungmin Lee, Department of Laboratory Medicine, Korea University Medical Center Guro Hospital, Korea.
Citation: Lee J., (2023), Severe Thrombocytopenia Developed from Bone Marrow Involvement of Chronic Disseminated Histoplasmosis in Immunocompetent Patient, Journal of Clinical and Laboratory Research. 6(2); DOI:10.31579/2768-0487/103
Copyright: © 2023, Jungmin Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 02 May 2023 | Accepted: 22 May 2023 | Published: 29 May 2023
Keywords: bone marrow involvement; histoplasmosis; thrombocytopenia; toxocariasis
Disseminated histoplasmosis among immunocompetent patients is rarely reported. When it evolves into chronic phase, it may be associated with clinically significant thrombocytopenia. In progressive disseminated histoplasmosis, thrombocytopenia and other cytopenia are hallmark of bone marrow involvement and therefore where there are refractory thrombocytopenia following anti-fungal medication and platelet transfusion, immune thrombocytopenic purpura (ITP) should be considered.
Histoplasmosis is a common infection endemic in many regions of America, Asia, India and Africa, with sporadic cases also occurring throughout the world [1]. Disseminated disease is rare but can be fatal if untreated. H. capsulatum is a dimorphic fungus found in the temperate zones of the world; the mycelial form of H. capsulatum is found in the soil, especially in areas contaminated with bird or bat droppings [2]. Infections in endemic areas are typically caused by wind-borne spores emanating from point sources such as bird roosts, old houses or barns, or activities involving disruption of the soil such as farming and excavation [3]. There are a few case reports of this disease seen in immunocompetent individuals [4]. We hereby report a case of severe thrombocytopenia developed from chronic disseminated histoplasmosis reactivation in immunocompetent patient.
A 63-year-old (2021) Asian man presented with history of disseminated histoplasmosis and toxocariasis diagnosed in year 2014 (5 years ago) presented with the recurred symptoms and neutropenia. He was a missionary who visits República de Honduras, Guatemala, Nicaragua, El Salvador, Brazil, and Argentina in Central America periodically. He did not have any contact with the bats; however, he had lived in dusty places with mosquitos during his visit in Central America. In September 2013, the following symptoms started to appear: abdominal distention, fever, night sweating, and palpable mass on the neck.
He was admitted to a hospital in Korea in October 2014, and he underwent several tests. From percutaneous biopsy and colonoscopy, the adrenal gland showed extensive coagulation necrosis in the adrenal cortex with neutrophilic infiltration and numerous Periodic acid-Schiff (PAS) and Gomori methenamine silver (GMS) was positive fungal yeast, which was consistent with histoplasmosis diagnosis. His bone marrow biopsy revealed normocellular marrow (40-50%) with a few fungal yeast, lymphoid aggregates, and granulomas showing bone marrow involvement of histoplasmosis. Ileum biopsy showed chronic active ileitis, many fungal yeast and adrenal biopsy showed extensive coagulation necrosis of adrenal cortex, neutrophilic infiltration, and numerous fungal yeasts. He was diagnosed of subacute progressive disseminated histoplasmosis (PDH) involving adrenal glands, colon, and the bone marrow. He was treated with itraconazole and albendazole for 1 year and the symptoms of fever, loss of appetite, high fever, and weight loss had subsided.
After returning to Central America, he visited Hospital Centro Médìco Hondureño, in Barrio La ranja in January 2016. His other symptoms have resolved and the labs showed normal levels. He only developed skin lesions that was suspiciously due to fungal infection.
However, starting September in 2019, symptoms such as weight loss, malaise, and fatigue started to reappear. The complete blood count was as follows: hemoglobin (hb), 10.3 g/dL; white blood cells (WBC), 1.0×109/L; platelets, 161×109/L; with eosinophilia (28.1%) and monocities (35.4%). Abdominopelvic computed tomography (APCT) showed lymph node enlargement aggravated compared with APCT four years ago. Further hematological and chemistry studies revealed increased CRP level to 108.55 mg/L, increased serum Vitamin B12 1014 pg/mL, folate 3.7 ng/mL increased serum ferritin 451.40 ng/mL, increased serum haptoglobin 325 mg/dL. Toxocariasis Ab IgG was positive (1.984, cut off 1.233) and histoplasma Ab screen was (mycelia Ab <1>1:64). Bone marrow aspirate showed normal cellularity of 30-40% with eosinophilia 8%. M:E ratio was 2.5:1. Erythroid and granulocytic precursors were normal in number and normal in maturation. Blasts were counted up to 0.6% of marrow ANCs. Megakaryocytes were normal in number and in maturation. Abnormal or malignant cell clusters were found. After antifungal treatment, symptoms and abnormal lab results did not resolve.
A year after, in October 2020, symptoms still stayed persistent with oral lesions. The patient’s CBC revealed pancytopenia (Hb 9.9 g/dL – WBC 0.93 ×109/L - PLT 87×109/L) with normocytic normochromic anemia, markedly decreased leukopenia, relative eosinophilia (17%), severe neutropenia (ANC 170 /uL) and marked thrombocytopenia. Toxocariasis Ab was positive as well as histoplasmosis Ab which showed mycelial Ab below 1:8 and yeast Ab 1:8). Soluble Interleukin2 receptor (sCD25, HLH) was elevated up to 1091U/mL. CRP was increased up to 209.74 mg/L. For immunohistochemical study, CD117, CD34, CD163 were done for diagnosis. CD34 and CD117 were negative whereas CD163 was positive in normal histiocytes. For special stains, GMS was negative for microorganism. Bone marrow aspirate showed normocellular marrow with left shifted maturation. The quality of bone marrow aspirate was adequate. M:E ratio was 3.2:1. Erythroid precursors were relatively decreased in number and maturation. Granulocytic precursors were increased in number with marked left-shifted maturation (promyelocytes: 35.2% of marrow ANCs). Histiocytes were frequently observed with occasional hemophagocytosis [Figure 1]. No microorganism was observed. Megakaryocytes were normal in number and morphology. The biopsy specimen showed normocellular marrow (40-50%) for his age. Megakaryocytes were normal in number and nucleated cells were mostly hematopoietic cells. His diagnosis was chronic disseminated histoplasmosis by this time.
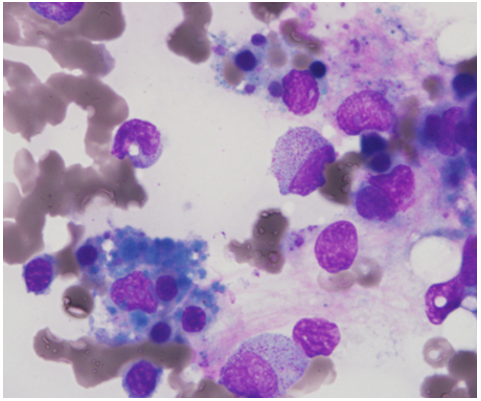
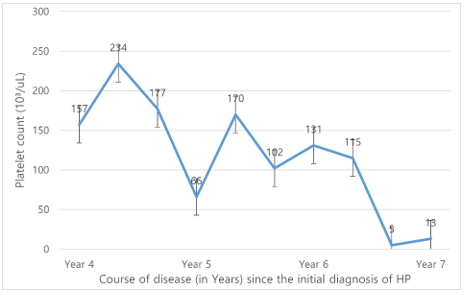
In March 2021, he had newly developed severe thrombocytopenia (platelet count 1.3×109/L). The complete blood count was hemoglobin, 13.3 g/dL; white blood cells, 1. 6×109/L; platelets, 1.3×109/L with eosinophilia 18.6% and monocities 20.0%. CRP level dropped to normal level. After anti-fungal treatment, thrombocytopenia did not improve. To avoid fatal complications of thrombocytopenia such as spontaneous bleeding, platelet transfusion was received. Bone marrow biopsy needs to be done to rule out histoplasmosis aggravation and if the results were consistent with immune thrombocytopenia purpura (ITP) then steroid treatment was to be considered. Because bone marrow aspirate did not show any aggravation or improvement [Figure 2]. Bone marrow failure due to previous infection was suspected.
The common presentation of histoplasmosis is fever with abdominal pain, weight loss, hepatosplenomegaly, lymphadenopathy; oral ulcerations, anemia and thrombocytopenia [4] In this case, while this patient was not overtly immunosuppressed, he progressed into subacute, then chronic PDH probably due continual exposal to a large inoculum [5]. In progressive disseminated histoplasmosis, thrombocytopenia and other cytopenia are hallmark of bone marrow involvement but rarely reports of patients. Presenting with isolated thrombocytopenia has been reported and in disseminated disease, thrombocytopenia purpura has been reported in association with pancytopenia [6]. Pathogenesis of thrombocytopenia in histoplasmosis according to Des Prez et al. has been found to be that the yeast from H. capsulatum activates the platelets resulting in serotonin release reaction and aggregation without the involvement of the complement pathway [7]. The plasma cofactors which are involved are IgG which is necessary for induction of release reaction [2, 4, 6, 8].
The chronic form of DH is characterized by an indolent course, focal lesions and an effective cell mediated immune response [9]. The subacute DH pursues a subacute, but relentless course and focal lesions in various visceral organs [9]. Chronic PDH slowly progresses and generally fatal and occurs mostly in older adults who is not immunosuppressed [8]. The patients have no obvious immunosuppression, but their macrophage clearly cannot effectively kill H. capsulatum [2, 4, 6, 8]. It is evident that the relationship between histoplasmosis and thrombocytopenia needs further study. However, it is important to stress that immune thrombocytopenia purpura should be considered in a systemic histoplasmosis.
None declared. This study was approved by the Korea University Guro Hospital Institutional Review Board (K2021-1310-001).
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
