
Review Article | DOI: https://doi.org/10.31579/2641-0419/038
1Jordan University of Science and Technology, School of Medicine, Irbid, Jordan.
2Ankara University, Faculty of Pharmacy, Ankara, Turkey.
3Department of Kinesiology and Nutritional Sciences, California State University, Los Angeles, USA.
4Institute of Cardiovascular Sciences, St. Boniface Hospital Albrechtsen Research Centre & Department of Physiology and Pathophysiology, Max Rady College of Medicine, University of Manitoba, Winnipeg, Canada
*Corresponding Author: Naranjan S. Dhalla, Institute of Cardiovascular Sciences St. Boniface Hospital Albrecheston Hospital Research Centre Winnipeg, Manitoba, Canada R2H 2A6
Citation: Nusier M., Ozcelikay AT., Shah AK., Dhalla NS. (2020) Role of Intracellular Ca2+-overload in Cardiac Dysfunction in Heart Disease. Clinical Cardiology and Cardiovascular Interventions, 3(2); Doi:10.31579/2641-0419/038
Copyright: © 2020 Naranjan S Dhalla, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 13 December 2019 | Accepted: 03 January 2020 | Published: 07 January 2020
Keywords: calcium overload; gene expression; sarcoplasmic reticulum; sarcolemma; myofibrils; subcellular organelles
Various heart diseases such as genetically-determined heart failure, acute myocardial infarction, ischemia-reperfusion injury and catecholamine-induced cardiomyopathies are associated with cardiac dysfunction, cellular damage, subcellular derangements and metabolic alterations. Since increase in myocardial Ca2+ is accompanied by these abnormalities, it is generally held that intracellular Ca2+-overload plays an important role in the pathogenesis of cardiac dysfunction as well as cellular and metabolic defects in different cardiovascular diseases. This view is supported by observations in hearts subjected to Ca2+-paradox, where reperfusion of Ca2+-free perfused hearts with Ca2+-containing medium was found to produce a marked increase in myocardial Ca2+-content, cellular damage and cardiac contracture. The intracellular Ca2+-overload in the heart has also been shown to produce mitochondrial Ca2+-overload, depress ATP production, release different toxic substances and induce cardiomyocyte apoptosis. By virtue of its ability to depress cardiac gene expression and increase proteolysis of sarcolemma (SL) sarcoplasmic reticulum (SR) and myofibrils (MF), the intracellular Ca2+-overload has been reported to reduce SL, SR and MF protein content and activities. Such remodeling of subcellular organelles is associated with dramatic alterations in Ca2+ -handling by SL and SR membranes as well as interaction of Ca2+ with MF for the impairment of cardiac function. Thus, it is evident that mitochondrial Ca2+-overload, and subcellular remodeling for Ca2+-handling defects are responsible for the occurrence of cardiac dysfunction, metabolic derangements and cellular damage during the development of heart disease.
Short Title: Myocardial Ca2+ and Cardiac Function
It is now well known that cardiac contraction and relaxation processes are determined by the coordinated functions of different subcellular organelles including sarcolemma (SL), sarcoplasmic reticulum (SR), mitochondria (MT) and myofibrils (MF) [1-6]. The SL proteins such as voltage-sensitive Ca2+-channels, store-operated Ca2+-channels, Na+- Ca2+ exchanger and Na+- K+ ATPase as well as SR proteins including Ca2+-release channels (ryanodine receptors) and Ca2+-pump ATPase play an essential role in the entry and regulation of Ca2+ in cardiomyocytes. On the other hand, MF Ca2+-stimulated ATPase and MT oxidative phosphorylation are involved in the generation of contractile force and ATP production, respectively. It is noteworthy to point out that Ca2+ is not only essential for determining the status of cardiac contractile function, but is also intimately involved in the maintenance of membrane permeability, cellular integrity, and cardiac gene expression [3,7-9]. Furthermore, various vasoactive hormones including catecholamines and angiotensin II have been demonstrated to exert marked effects on Ca2+-transport activities in cardiomyocytes [4,10,11]. Thus, defects in any of the components of subcellular organelles can be seen to induce Ca2+-handling abnormalities and contractile dysfunction of the heart [3,9].
Since the identification of Ca2+-overload as a new principle for the pathophysiology of cardiac dysfunction [12-14], several diseases including cardiomyopathies due to high levels of circulating catecholamines [15-20], genetically-determined heart failure [21-25] as well as ischemic heart disease (acute myocardial infarction [26-30] and ischemia-reperfusion injury [31-35]) have been shown to be associated with the development of intracellular Ca2+-overload. It is generally assumed that impaired cardiac performance and functional derangement of subcellular organelles in different diseases are the consequence of intracellular Ca2+-overload. It should also be pointed out that there are other pathophysiologic mechanisms including oxidative stress and myocardial inflammation, which have been proposed to induce cardiac dysfunction and cellular abnormalities during the development of heart disease [36-40]. However, in this article we have attempted to highlight the evidence that intracellular Ca2+-overload plays a critical role in the genesis of metabolic and cellular defects as well as subcellular remodeling for the development of cardiac dysfunction in the heart. Furthermore, the present review is focussed on discussion of events for the occurrence of intracellular Ca2+-overload in cardiomyocytes and its consequences for inducing myocardial abnormalities.
Mechanisms for the Development of Intracellular Ca2+-overload
Although high levels of circulating catecholamines are known to produce intracellular Ca2+-overload, several mechanisms have been proposed to underlie this phenomenon [9,16,18,20]. These include activation of both α-and β-adrenoceptors, stimulation of SL Ca2+-channels, depression in SL Na+-Ca2+-exchanger and SL Ca2+-pump ATPase as well as oxidation of catecholamines and formation of oxyradicals. It is pointed out that interventions which reduce the entry of Ca2+ as well as prevent the oxidation of catecholamines and development of oxidative stress have been shown to attenuate the catecholamine-induced intracellular Ca2+-overload [9,12,16,18]. Furthermore, the occurrence of intracellular Ca2+-overload in genetically-determined cardiomyopathy has been attributed to the activation of sympathetic nervous system and increase in Ca2+-influx as well as the depression of SL Na+-K+ ATPase and increase in intracellular Na+ [9,21]. Agents such as Ca2+-antagonists which prevent the entry of Ca2+ in the heart have been reported to exert beneficial effects in cardiomyopatheic animals by reducing the development of intracellular Ca2+-overload[9,22,25].
Several studies have been conducted to demonstrate mechanisms for the occurrence of intracellular Ca2+-overload due to acute coronary occlusion as well as ischemia-reperfusion injury [9,27,28,30, 33-35]. It has been shown that the lack of oxygen in the ischemic myocardium results in acidification of the cytoplasm which promotes SL Na+-H+ exchange and subsequent entry of Ca2+ upon stimulation of Na+-Ca2+ exchange system. Lack of oxygen is also known to increase membrane permeability for Ca2+ due to incorporation of free fatty acids and other lipid metabolites in the SL membrane. On the other hand, ischemia-reperfusion injury has been associated with the release of norepinephrine from the adrenergic nerve endings for increasing the entry of Ca2+ in addition to promoting the development of oxidative stress. These changes are known to cause the occurrence of intracellular Ca2+-overload as a consequence of their dramatic effects on the SL membrane [9,27,33,37,38]. Several other vasoactive interventions and proinflammatory agents have also been shown to produce Ca2+-handling abnormalities in cardiomyocytes [39,40]. It may be noted that reperfusion of the Ca2+-depleted heart with Ca2+ containing medium has been shown to exhibit Ca2+-paradox and provide a direct evidence for the occurrence of intracellular Ca2+-overload [9, 41-44]. A massive increase in myocardial Ca2+content due to stimulation of Na+-Ca2+ exchanger in this experimental model was shown to be prevented when perfusion of the heart with Ca2+-free medium was carried out in the presence of low Na+ [42,43]. High concentrations of Ca2+-antagonists were also found to attenuate the increase in myocardial Ca2+ in the Ca2+-paradoxic heart by their action on the SL Na+-Ca2+ exchange activity [44]. Thus, the Ca2+-paradoxic heart is considered to form an excellent model for studying the effects of intracellular Ca2+-overload [42,43].
Cardiac Dysfunction and Cellular Damage
Reperfusion of the Ca2+-depleted hearts with Ca2+-containing medium was found to result in loss of contractility, development of contracture, damage to ultrastructure and leakage of intracellular enzymes from the myocardium [41, 45-48]. The paradoxical effects of Ca2+-deprived hearts were reported to occur in different species [49] and were similar to those seen during the development of oxygen- paradox in normal hearts [50]. The Ca2+-paradox phenomenon was shown to be associated with irreversible changes in the surface electrical activity [41] and a marked increase in the left ventricular end-diastolic pressure (LVEDP) [41,42,51-53]. The occurrence of intracellular Ca2+-overload and the increase in LVEDP (Table 1) as well as the development of cardiac contracture in the Ca2+-paradoxic heart were found to be dependent upon the concentration of Ca2+ in the reperfusion medium [42,53,54].
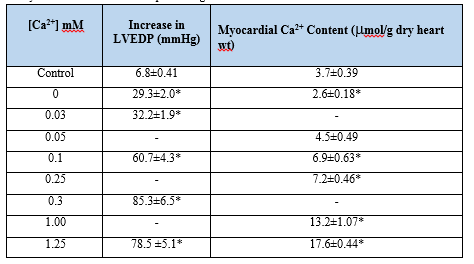
Although some investigators failed to demonstrate Ca2+-paradox associated changes in isolated cardiomyocytes [55], others have shown these alterations upon successive exposure of cardiomyocytes to Ca2+-free medium and Ca2+-containing medium [48, 56-58]. Nonetheless, ischemic preconditioning has been observed to attenuate the Ca2+-paradox associated increase in LVEDP, depression in the left ventricular developed pressure and leakage of myoglobin from the heart [59]. The presence of low Na+ during perfusion of the heart with Ca2+-free medium was also found to prevent the development of cardiac dysfunction and the occurrence of intracellular Ca2+-overload upon reperfusion [41-43].
The ultrastructural changes in the Ca2+-deprived and reperfused hearts included swelling of mitochondria and sarcotubular system, occurrence of contractile bands, and partial separation of the intercalated disc as well as basement membrane from sarcolemma [41,43,45,60]. The alterations in ultrastructure of the myocardium were dependent upon the concentration of Ca2+ in the reperfusion medium [41,60] and were attenuated by reducing the concentration of Na+ during the Ca2+-free perfusion phase [41]. These ultrastructural changes are similar to those seen in the ischemic heart disease [27-28] and may be a consequence of increased activities of cardiac lysosomal hydrolases [61], different intracellular proteases [35] and phospholipases [62]. Although the occurrence of autophagy has been reported in ischemia-reperfused hearts and myocardial infarction [27,28], no information regarding autophagic changes in the Ca2+-paradoxic heart is available at present. It is pointed out that the activation of NFκB and increased production of TNF-α have also been reported to cause cardiac injury due to intracellular Ca2+-overload [63]. Furthermore, the occurrence of cell death (apoptosis) in the Ca2+-paradox heart has been associated with the activation of mitogen-activated protein kinases (p38 and ERK) as well as different apoptotic signal transduction pathways [64]. Thus, the development of cardiac dysfunction and cellular damage due to intracellular Ca2+-overload appears to be occurring as a consequence of complex and diverse mechanisms.
Mitochondrial Ca2+-overload and Energy Depletion
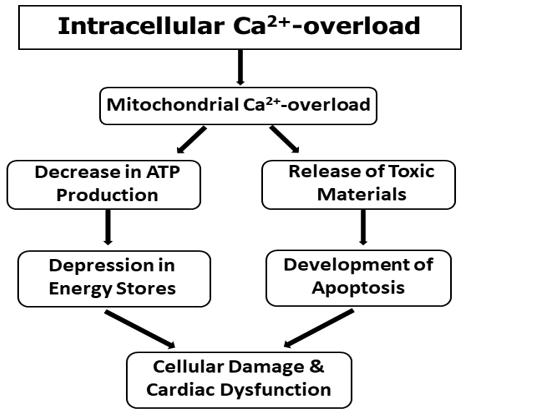
It is now well known that intracellular Ca2+-overload in the heart results in the development of mitochondrial Ca2+-overload and defects in energy production [9,47,65]. Although low concentrations of Ca2+ are required for the stimulation of mitochondrial oxidative phosphorylation, high concentrations of Ca2+ have been shown to impair the mitochondrial function for ATP production [9,53,65, 66]. Perfusion of hearts with Ca2+-free medium followed by reperfusion with Ca2+-containing medium for the induction of intracellular Ca2+-overload was found to be associated with depressed mitochondrial state 3 respiration, respiratory control index, ADP/O ratio and oxidative phosphorylation without any changes in state 4 respiration [53,67]. These alterations were prevented when the reperfusion was carried out at low concentrations (0.1-0.5 mM) of Ca2+ but were not affected by different antioxidants [55]. The impaired mitochondrial function in the Ca2+-paradoxic heart has been associated with elevated levels of citric acid cycle intermediates and is considered to be due to defects in mitochondrial membrane potentials [68,69].
A dramatic decrease in high -energy phosphate stores in the heart has been shown to occur upon the induction intracellular Ca2+-overload [67,70,71]. It may be noted that Ca2+-binding and Ca2+-uptake activities of mitochondria, isolated from the Ca2+-paradoxic hearts, were found to be increased [72]. Such a change in the mitochondrial Ca2+-transport activity was suggested to contribute towards the occurrence of mitochondrial Ca2+-overload as it was attenuated when the perfusion with Ca2+-free medium was carried out in the presence of low Na+ [72]. It is also pointed out that mitochondrial Ca2+-overload may release several cytotoxic substances, which may also serve as signals for inducing apoptosis in the Ca2+-paradoxic hearts [64]. Thus, it appears that mitochondrial Ca2+-overload may be involved in cardiac dysfunction and cellular damage in the heart by depressing the high energy phosphate stores as well as inducing apoptosis in the myocardium. A schematic representation of these events is shown in Figure 1.
Subcellular Defects and Ca2+-handling Abnormalities
While the SL membrane is concerned with influx and efflux of Ca2+ for maintaining Ca2+-homeostasis in cardiomyocytes, the SR tubular system is involved in raising and lowering the concentration of Ca2+, whereas the interaction of Ca2+ with MF proteins determines the contractile status of the myocardium [3,4]. Reperfusion of Ca2+-deprived hearts with Ca2+-containing medium has been shown to exert profound effects on the activities of different subcellular organelles (Table 2) [73-75].
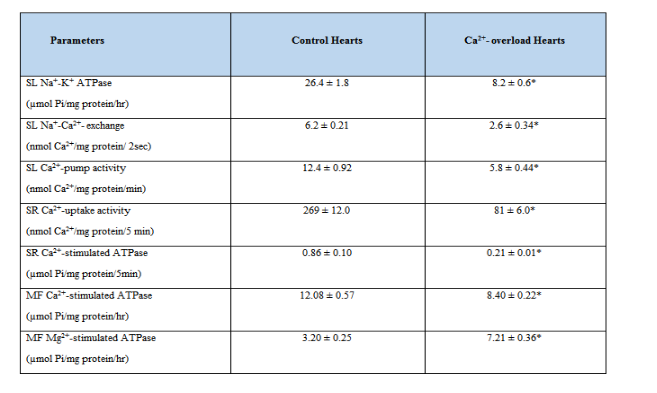
Depressions in the SL Na+-K+ ATPase, SL Na+-Ca2+ exchanger and SL Ca2+-pump ATPase activities in the Ca2+-paradoxic heart can be seen to contribute towards the occurrence of intracellular Ca2+-overload in cardiomyocytes [73,76,77]. These SL defects were attenuated when the perfusion with Ca2+-free medium was carried out in the presence of low Na+ (35mM) or at low temperature (210C) [42,78]. On the other hand, the density of SL Ca2+-channels was increased upon subjecting the heart to Ca2+-paradox and this change was also attenuated by carrying out the perfusion with Ca2+-free medium in the presence of low Na+ or at low temperature [79]. Furthermore, alterations in the SL membrane were also apparent because the activities of β-AR – G-protein – adenylyl cyclase complex were observed to be increased [80] and the activity of SL Ca2+/Mg2+-ecto ATPAse was decreased [81] in the Ca2+-paradoxic heart. Although the status of SL store-operated Ca2+-channels [6] in the Ca2+-paradoxic heart has not be determined, their participation in inducing intracellular Ca2+-overload can not be ruled out at present.
The induction of Ca2+-paradox in the heart upon perfusion with Ca2+-free medium followed by Ca2+-containing medium was seen to be associated with marked depression in the SR Ca2+-uptake and release activities [72,74]. These changes in Ca2+-handling by SR were dependent upon the concentration of Ca2+ in the reperfusion medium and were attenuated when the perfusion with Ca2+-free medium was carried out in the presence of low Na+ or at low temperature. Although MF Ca2+-stimulated ATPase activity was not altered during the initial (5 min) reperfusion phases of Ca2+-paradox development [67], reperfusion of Ca2+-deprived hearts with Ca2+-containing medium for 10 min was found to depress the MF Ca2+-stimulated ATPase activity and increase the MF Mg2+-ATPase activity [75]. These alterations were associated with degradation of MF α -myosin heavy chain and troponin T proteins in the Ca2+-paradoxic hearts. The activation of proteases such as calpain by elevated levels of intracellular Ca2+ in cardiomyocytes is considered to be involved in alterations of the SL, SR and MF activities upon reducing their protein content [35]. These events for inducing subcellular defects due to the occurrence of intracellular Ca2+-overload in the Ca2+-paradoxic hearts are shown in Figure 2.
It should be recognized that Ca2+-handling abnormalities in SL and SR due to intracellular Ca2+-overload may also be induced by changes in the phospholipid composition of these membranes [62]. It is also noteworthy that similar Ca2+-handling defects have also been observed in heart failure and ischemic heart disease [27,28,82-85].
Alterations in cardiac Gene Expression
In view of the role of cardiac gene expression in maintaining the function of different subcellular organelles in the heart [27,28, 85], it has been suggested that subcellular remodeling in the Ca2+-paradoxic heart may be due to changes in gene expression for different subcellular proteins [9,73,74]. Accordingly, subcellular remodeling due to intracellular Ca2+-overload may be occurring as a consequence of both the activation of calpain and the depression in mRNA levels for different cardiac genes (Figure 2).

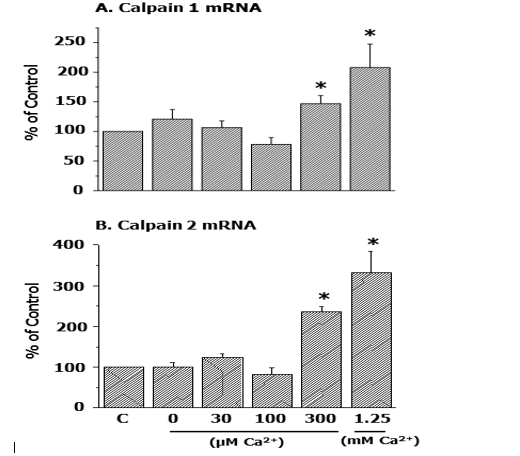
In this regard, it may be noted that the increase in mRNA levels for both calpain 1 and calpain 2 in the Ca2+-paradoxic heart was found to be dependent on the concentration of Ca2+ in the reperfusion medium (Figure 3) [54].
Furthermore, it was demonstrated that depressions in gene expression for SL Na+-Ca2+ exchanger as well as different isoforms of SL Na+- K+ ATPase protein due to Ca2+-paradox were dependent upon the concentration of Ca2+ in the reperfusion medium (Figure 4) [54].
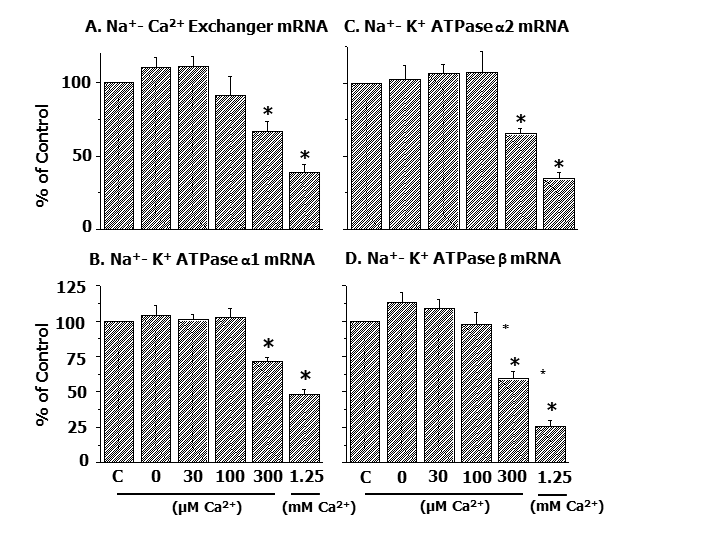
Likewise, alterations in mRNA levels for SR Ca2+-pump protein and Ca2+-release channels as well as MF α- and β- myosin proteins in the Ca2+-paradoxic heart were observed to be dependent upon the concentration of Ca2+ in the reperfusion medium (Figure 5) [54].
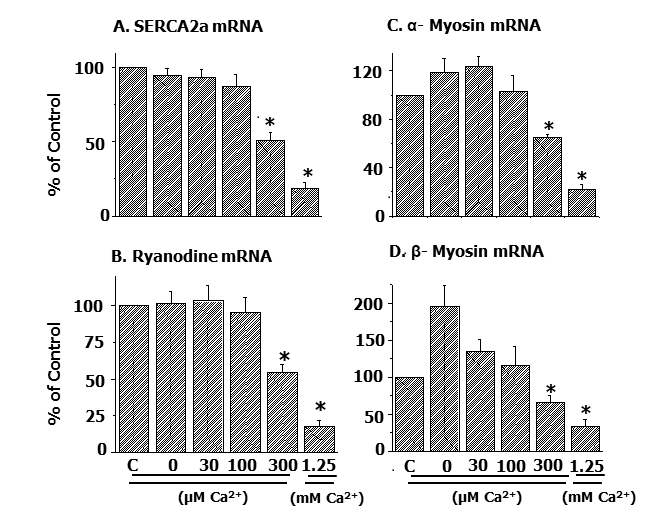
These observations provide evidence for a defect in the formation of subcellular proteins resulting in subcellular remodeling due to intracellular Ca2+-overload. Thus, cardiac genes can be seen as excellent molecular targets for the development of novel interventions for the improved therapy of heart disease.
From the forgoing discussion, it is evident that two major mechanisms, namely energy depletion due to mitochondrial Ca2+-overload and subcellular remodeling due to increased proteolysis and reduced gene expression, are likely to explain the development of cellular damage, metabolic alterations and cardiac dysfunction due to intracellular Ca2+-overload. It is emphasized that the occurrence of intracellular Ca2+-overload in heart disease may become apparent due to increase in Ca2+ entry as a consequence of depressions in SL Na+-K+ ATPase and Na+-Ca+ exchange activities as well as increase in Ca2+-channel density in the SL membrane. Depressions in SL Ca2+-pump ATPase as well as SR Ca2+-uptake and SR Ca2+-release activities in heart disease can also be seen to participate in the development of intracellular Ca2+-overload. Since the observed changes in subcellular Ca2+- handling due to intracellular Ca2+-overload are similar to those seen in failing hearts and thus may be responsible for the development of cardiac dysfunction in different types of heart types of heart disease. It may be noted that the SL and SR defects during the development of heart disease are also induced by prolonged exposure of the heart to elevated levels of vasoactive hormones such as catecholamines and angiotensin II in the circulation. The accumulation of Ca2+ by mitochondria under conditions of intracellular Ca2+-overload may be beneficial at initial stages but the resultant mitochondrial Ca2+-overload can be seen to impair ATP production and promote the development of cellular damage. Thus, different interventions which can attenuate the Ca2+ entry into cardiomyocytes, reduce the occurrence of mitochondrial Ca2+-overload, inhibit the activation of proteases and promote cardiac gene expression can be seen to exert beneficial effects in preventing the development as well as progression of heart disease.
The infrastructure support for this project was provided the St. Boniface Hospital Research Foundation, Winnipeg, Canada.
The authors declare that there was no conflict of interest.
From the forgoing discussion, it is evident that two major mechanisms, namely energy depletion due to mitochondrial Ca2+-overload and subcellular remodeling due to increased proteolysis and reduced gene expression, are likely to explain the development of cellular damage, metabolic alterations and cardiac dysfunction due to intracellular Ca2+-overload. It is emphasized that the occurrence of intracellular Ca2+-overload in heart disease may become apparent due to increase in Ca2+ entry as a consequence of depressions in SL Na+-K+ ATPase and Na+-Ca+ exchange activities as well as increase in Ca2+-channel density in the SL membrane. Depressions in SL Ca2+-pump ATPase as well as SR Ca2+-uptake and SR Ca2+-release activities in heart disease can also be seen to participate in the development of intracellular Ca2+-overload. Since the observed changes in subcellular Ca2+- handling due to intracellular Ca2+-overload are similar to those seen in failing hearts and thus may be responsible for the development of cardiac dysfunction in different types of heart types of heart disease. It may be noted that the SL and SR defects during the development of heart disease are also induced by prolonged exposure of the heart to elevated levels of vasoactive hormones such as catecholamines and angiotensin II in the circulation. The accumulation of Ca2+ by mitochondria under conditions of intracellular Ca2+-overload may be beneficial at initial stages but the resultant mitochondrial Ca2+-overload can be seen to impair ATP production and promote the development of cellular damage. Thus, different interventions which can attenuate the Ca2+ entry into cardiomyocytes, reduce the occurrence of mitochondrial Ca2+-overload, inhibit the activation of proteases and promote cardiac gene expression can be seen to exert beneficial effects in preventing the development as well as progression of heart disease.
The infrastructure support for this project was provided the St. Boniface Hospital Research Foundation, Winnipeg, Canada.
The authors declare that there was no conflict of interest.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
