
Review Article | DOI: https://doi.org/10.31579/2690-8816/107
*Corresponding Author: Salma M AlDallal, Department of Hematology, Amiri Hospital, Kuwait City, Kuwait.
Citation: Salma M AlDallal. (2023). Renal Manifestations in Patients with Sickle Cell Disorder. J Clinical Research Notes, 4(3); DOI:10.31579/2690-8816/107
Copyright: © 2023, Salma M AlDallal. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 23 February 2023 | Accepted: 11 March 2023 | Published: 22 March 2023
Keywords: kidney papillary necrosis; proteinuria; kidney failure; renal insufficiency; anemia; biomarkers; SCN; SCD; renal medulla
In recent years, sickle cell nephropathy (SCN) has gained recognition for its distinct symptoms, prognoses, and risk factors. It is identified by sickled erythrocytes in the renal medullary arteries leading to a range of renal consequences. In addition to proteinuria associated with glomerular damage, renal papillary necrosis, distal nephron failure, and renal acidification defect, SCN also involves other renal symptoms that can progress to end-stage renal disease. Age is one of the determining factors in the development of proteinuria in sickle cell disease (SCD), which begins as microalbuminuria in adolescence and escalates to macroalbuminuria. Chronic kidney disease develops due to interactions between various processes involving the tubular, vascular, interstitial, and glomerular compartments of the kidney in some SCD patients who experience proteinuria. Early detection of the disease by non-invasive kidney damage biomarkers and incorporating them into clinical practice will enable understanding the syndrome’s underlying mechanisms. It will help create more efficient procedures for preventive and therapeutic approaches to SCD management.
Sickle cell nephropathy (SCN) describes various sickle cell diseases (SCD) accompanied by renal symptoms. Kidney disease is among the most prevalent and dangerous consequences of SCD. The latent manifestation of SCN since infancy is responsible for mortality in adults [1,2]. SCN is characterized by sickled erythrocytes in the renal medulla, which causes reduced medullary blood flow, microinfarcts, ischemia, and papillary necrosis in the kidneys. Disturbances in tubular and glomerular function brought on by these pathologic alterations impact the control of blood pressure and the metabolism of water and electrolytes [3].
SCD is one of the most prevalent inherited hematologic diseases worldwide. Sickle cell anemia (SCA) is caused due to homozygous mutation in the β-globin genes. Regarding its renal symptoms, SCA is the most severe variant of SCD [4]. Over 30000 infants worldwide are born with SCD, an autosomal recessive hemoglobinopathy. About 5–18% of SCD cases present renal dysfunction. Nephrotic syndrome, glomerular disorders, and sickle cell anemia are linked. Approximately 40% of fatal adult SCD cases result from renal failure [5].
Hemolysis and vaso-occlusion are the primary pathogenic factors in SCD, which cause endothelial vasculopathy and dysfunction,
ischemia/reperfusion injury, hypercoagulability, oxidative stress, nitric oxide deficit, activation of platelet, and enhanced neutrophilic adhesion. Consequently, both acute and chronic symptoms potentially harm multiple organs. Chronic anemia and painful bone crises are frequent. Acute chest syndrome, pulmonary hypertension or fibrosis, sepsis, functional asplenia, retinopathy, cerebral infarcts and stroke, avascular necrosis of upper extremities, lower-extremity ulcers, or SCN itself are additional manifestations [1,6].
A wide range of renal disorders is defined under SCN. The renal medulla, which is supplied by the vasa recta capillaries, is the primary site of kidney damage. Chronic sickling of blood cells underlies several mechanisms for kidney damage in SCN that are linked to hypertonicity, low pH, and low O2 tension of the renal medulla. These factors encourage the hemoglobin polymer formation in the sickled red cells, which increases interstitial edema, functional venous engorgement, and blood viscosity. This puts the microcirculation of the kidneys at risk of ischemia or infarction [3].
Sickling, in turn, affects the vasculature in several ways, including increased adhesion of red blood cells (RBCs) to the vascular wall, increased inflammation, increased vascular tone (i.e., causing vasoconstriction, as free hemoglobin from hemolysis sequesters nitric oxide and activating platelets and coagulation factors (Figure 1) [4].
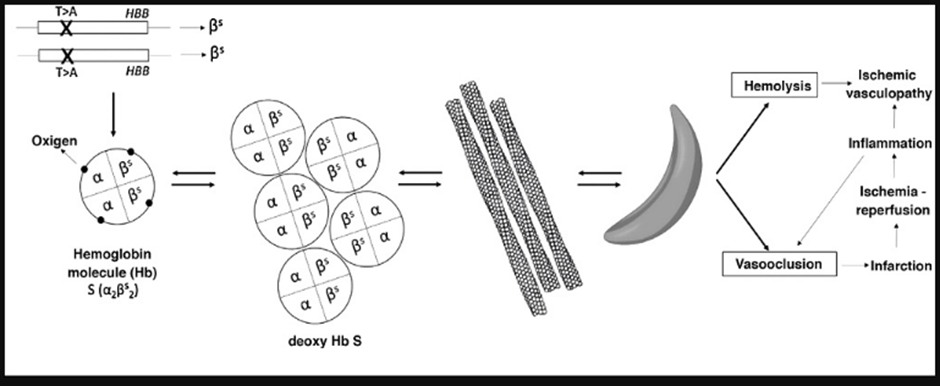
Figure 1: Sickle cell disease: pathophysiology
Clinical manifestations of SCN (Figure 2)
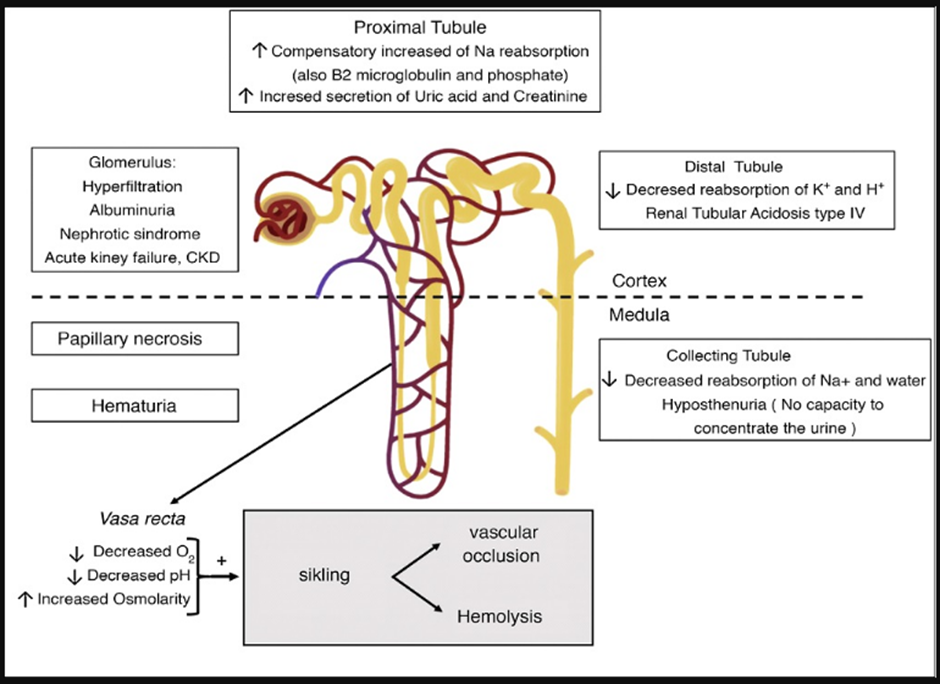
Figure 2. Sickle cell disease’s renal manifestations [1]
Hematuria and renal papillary necrosis (RPN)
Patients with SCD frequently experience hematuria. Those with sickle cell trait may experience this condition twice as frequently [10-12]. 13–30% of SCD patients report having microscopic or macroscopic hematuria, positively correlated with male gender and increased age [13]. It results from microinfarctions and vaso-occlusive events that damage the renal papillae. Greater venous pressure is applied to the left renal vein because it is longer and wedged between the aorta and the superior mesenteric artery. This causes relative hypoxia in the renal medulla and encourages sickling [1,14]. The infarctions could become severe, leading to renal papillary necrosis (RPN). Though rare, it can occur in about 30–40% of homozygous individuals (Hb S/S). Even hydronephrosis can appear clinically with asymptomatic hematuria, fever, and acute obstructive renal failure [14].
Renal ultrasonography can be used to diagnose RPN, even though CT scans are more accurate and might require diagnosis confirmation [15]. If a patient with SCD and hematuria does not have hypercalciuria, RPN is first seen on ultrasound as an enhanced medullary pyramids’ echogenicity. Following that, medullary pyramids might develop calcification encircling the renal pelvis or exhibit echogenicity problems in any pyramid due to the loss of the papilla [1,14].
Some individuals with severe or resistant hematuria have responded well to desmopressin treatment. A modest dose and caution should be used with antifibrinolytic medications like tranexamic acid or aminocaproic acid due to the possibility of thrombosis [1]. In rare instances, the afflicted renal segment may need to be embolized specifically [16]. Iron replacement may be required in situations when the hematuria is chronic.
Hyposthenuria
Hyposthenuria is a common observation in SCD and is characterized by the incapability of concentrating urine more than 450 mOsm/kg when water is restricted. It occurs in people with genotype S/S or S/-thalassemia in those with sickle cell trait at around the age of ten, and it is irreversible after that [4,10].
In addition to increased dehydration risk, exacerbated by inadequate water consumption, or higher extrarenal losses, hyposthenuria frequently manifests as polyuria, nocturnal enuresis, and nocturnal enuresis, favoring the onset of vaso-occlusive crises. Consequently, people with sickle cell disease are advised to drink 3/4 liter of fluids every day [15]. The level of hyposthenuria is inversely correlated with Hb S concentration, and therapy with hydroxyurea for 24 months is found to improve urine concentrating ability in young children as compared to placebo [17].
Tubular acidosis
Sickle formation first appears in the medulla’s vasa rectus region resulting in the tubule not being flushed out by the circulation after sodium reabsorption. It prevents free water from being absorbed again and causes medullary congestion. The most extended Henle loops, which start from the juxtamedullary nephrons and descend into the deep medulla and the papilla, are lost over time due to recurrent cycles of infarction and ischemia, which also cause collaterals to arise and the destruction of the vasa recta [9,13].
Patients with SCD may experience incomplete type IV renal tubular acidosis, accompanied by hyposthenuria, due to a deficiency in the distal secretion of hydrogen ions and potassium. Although the precise pathogenic process is not known, this might be caused by a reduction in the collecting duct’s electrochemical gradient due to reduced medullary blood flow, which causes hypoxia. Since the Henle loop’s deepest point suffers the most damage, it is pretty uncommon to have a substantial failure in urine acidification, which results in incomplete acidosis. Unless additional factors impair the kidney’s compensatory mechanisms, like the use of medicines that inhibit the renin-angiotensin-aldosterone system, beta-blockers, or potassium-sparing diuretics, hyperkalemia is rare and moderate [1,14].
Hyperfiltration
In individuals with SCD, albuminuria often occurs before hyperfiltration or an elevated GFR (in women >130 ml/min/1.73 m2 and >140 ml/min/1.73 m2 in males), which may be present from early infancy. Studies report that about half of homozygous adult patients showed hyperfiltration, and in half of them, it was linked to albuminuria [9]. Patients with SCD have lower filtration fractions than healthy individuals due to increased renal plasma flow, which is even greater than GFR. This phenomenon could be due to the loss of the juxtamedullary nephron, which is selective since they have a greater filtration percentage than cortical nephrons. It is most likely connected to the production of vasodilator chemicals, including prostaglandins, in the renal medulla due to ischemia. The vasodilatation will result in a reduction in renal vascular resistance and an increase in renal blood flow. A decrease in GFR has been linked to indomethacin’s inhibition of prostaglandin production. As a result, the kidney serves as an example of the “perfusion paradox,” which occurs in SCD in a single organ. Hyperperfusion is characteristic of the systemic circulation and some regional circuits but is common in the microvascular beds due to vaso-occlusion by Hb S [4,7].
Enhanced proximal tubular operation
The adaptive improvement in proximal tubular performance and the rise in sodium reabsorption are connected to the rise in GFR. Reports show high uric acid and creatinine secretion, increased reabsorption of 2-microglobulin and phosphate, and enhanced maximum transport of para-amino hippuric acid [4,7]. Due to increased oxidative stress, higher salt reabsorption necessitates oxygen consumption and favors tubular damage. Additionally, increasing oxygen demand will worsen hypoxia, encourage illness, and exacerbate renal injury [2].
Proteinuria
According to reports, 17–33% of SS patients have proteinuria [18]. The glomerular endothelium and podocytes may be damaged due to the hemodynamic alterations previously mentioned, leading to proteinuria [10,19] and tubulointerstitial injury. The most prevalent glomerulopathy in SCD, localized and segmental glomerulosclerosis, is brought on by these adhesions. It is observed in 39% or more of kidney biopsies taken from individuals with SCD who also have proteinuria and/or kidney failure. Less often seen histological patterns include thrombotic microangiopathy and membranoproliferative glomerulonephritis. Hemosiderin deposits in the tubular cells and hypertrophy of the glomeruli are virtually always observed [20]. In the first thirty years of life, up to 27% of people have albuminuria, while up to 68% of older patients have it. However, only 4% of SCD patients had proteinuria in the nephrotic range, which is quite uncommon [11,12]. A 5-year research found that more than 500 mg/g Cr albuminuria was linked to the development of chronic renal disease (CKD) [6,21,22]. In SCD patients with albuminuria >100 mg/mmol (884 mg/g), it is advised to begin therapy with ACEI or ARA-II [23].
Reduction in Blood pressure
According to a meta-analysis, Hb S/S genotype individuals have a significantly lower mean diastolic and systolic blood pressure than healthy controls of similar age and gender than healthy controls [6]. In addition, renal failure and albuminuria below 300 mg/g creatinine appear to be linked to persons with sickle cell anemia who are resistant to developing hypertension [24]. According to a study, relative hypertension in SCD is described as having a systolic blood pressure (SBP) between 120 and 139 mmHg or a diastolic blood pressure (DBP) between 70 and 89 mmHg. It is linked with a greater risk of pulmonary hypertension (PH) and renal impairment [25]. Keeping blood pressure under 130/80 mmHg is recommended based on the advantages shown in the general population [6].
Acute and chronic kidney failure
In individuals with SS illness, acute renal failure has been characterized as a component of multiorgan failure syndrome (MOFS) [12]. Between 4-10% of hospitalized SCD patients experience acute renal failure. A 15% drop in the reversible creatinine clearance has been observed during pain-related vaso-oclusive crises [26]. Rhabdomyolysis, volume depletion, infections, or non-steroidal analgesic use predisposes acute renal failure [4]. Like iron overload, SCD frequently develops and requires chelating treatment, while deferasirox is the most used medication. A dosage reduction should be considered if the serum creatinine level rises. This medication can potentially cause a dose-dependent, reversible rise in serum creatinine [27].
In a study of 410 SCD individuals between the ages of 2 and 21, 26.5% had CKD stage 1, 14.5% had stage 2, and 11.6% had stage 2 of the disease [28]. Another study found that CKD was the cause of 10.5% of fatalities in adult SCD patients. CKD was the primary cause of death in 43% of instances in homozygous individuals older than 60 [29]. Our country’s sole medication for SCD, hydroxyurea, is processed in the kidneys. Hence the dose must be changed following renal function. Additionally, it must be given after the hemodialysis session because hemodialysis removes it.
Diagnostic considerations
As serum creatinine doesn’t rise until the very end of SCN, renal problems are challenging to detect in the early stages. Only under severe proteinuria, abnormal GFR and increased serum creatinine levels appear. There is no agreement on the lab tests that should be performed to screen for sickle cell nephropathy or on the frequency at which they should be performed. However, it is advised to check renal function at least once a year starting at age 10, though it may be helpful to do so sooner [6]. It would be wise to use a urinary strip to check the morning’s first urine and measure the following: total proteins, sodium, creatinine, albumin, osmolality, and potassium; urinary creatinine/protein and creatinine/albumin indices; bicarbonate and cystatin C in blood; osmolality; osmolality; albumin; osmolality; and total proteins; (venous). A minimum of two consecutive urine samples, including one in the morning, are required to establish the presence of albuminuria [6].
Referring patients to nephrology is one strategy if the protein-creatinine ratio is greater than 50 mg/mmol (442 mg/g), there is proof of chronic microscopic hematuria, the estimated GFR is declining annually by more than 10%, or the GFR is predicted to be less than 60 ml/min/1.73 m2. One expert compiling every case at each center might be suggested. Knowing the signs of SCD is essential for the nephrology specialist. It is necessary to consider different causes of proteinuria or hematuria. The value of renal biopsy has not been evaluated, even though it should be performed in patients with nephrotic syndrome; therefore, each case must be decided on an individual basis [1,13,23].
Sickle cell disease refers to a set of linked hemoglobinopathies in which the sickle hemoglobin mutation is co-inherited with another beta globin mutation, resulting in sickling and vaso-occlusion. Sickle cell disease causes several renal syndromes and diseases, as well as significant changes to renal structure and function. Sickle cell nephropathy has elevated to a significant research focus due to its catastrophic consequences on patient mortality, morbidity, and quality of life. Despite the diligent efforts of researchers in the field, approaches to management and diagnosis have not yet been optimized. As a result, the discovery of early, non-invasive kidney injury biomarkers and their incorporation into clinical practice will aid in discovering the mechanisms underlying the emergence of renal syndromes, thereby assisting in establishing more efficient prevention and treatment methods.
The author confirms sole responsibility for the following: study conception and design, data collection, analysis and interpretation of results, and manuscript preparation.
Not Applicable.
None.
None
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
