
Research Article | DOI: https://doi.org/10.31579/2578-8868/215
1 International Neuroscience Institute (INI), Rudolf-Pichlmayr-Str. 4, D-30625 Hannover, Germany
*Corresponding Author: Gerhard Franz Walter, International Neuroscience Institute (INI) Rudolf-Pichlmayr-Str. 4 D-30625 Hannover / Germany.
Citation: Gerhard Franz Walter (2021). Range of Clinical Neuro-Ophthalmopathology: more than Tumors. J. Neuroscience and Neurological Surgery. 9(5); DOI:10.31579/2578-8868/215
Copyright: © 2021 Gerhard Franz Walter, This is an open-access article distributed under the terms of The Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 21 October 2021 | Accepted: 19 November 2021 | Published: 24 November 2021
Keywords: ophthalmopathology; neuropathology; clinical neurosciences; diagnostic procedures
The importance of close co-operation of ophthalmologists, neurologists, neurosurgeons and neuro-ophthalmopathologists as well in clinical as in scientific settings is underlined. Typical neuro-ophthalmopathological examples of frequent and rare cases from pathology of the eyelid, the cornea, intraocular tumors, ocular trauma and tumors of the orbit are presented as well as systemic pathologies such as inflammatory diseases of the eye, phakomatoses, malformations and mitochondrial disorders in which the histopathological investigation by neuro-ophthalmopathologists may contribute to diagnosis and therapeutic decision-making. The chosen examples should provide a narrow focus on some clinical queries answered by neuro-ophthalmopathology and exemplify methodological options, but – in the given frame – cannot represent the full range of eye pathology. Relevant literature is included for further reading. Ophthalmologists, neurologists and neurosurgeons should be aware from the additional value of a concise neuro-ophthalmopathological diagnosis for the optimal treatment of eye diseases and should insist in a professional neuro-ophthalmopathological investigation whenever and wherever possible.
Aim of this overview is to interest ophthalmologists and clinical neuroscientists in neuro-ophthalmopathology and to make them aware that a close co-operation with neuro-ophthalmopathologists on clinical as well as scientific questions can valuably contribute to the improvement of diagnostic and therapeutic decisions. The more precise the clinician’s question to the neuro-ophthalmopathologist – it does not matter whether she or he is general pathologist or neuropathologist – is formulated, the more precise the answer that may be given.
It is well known to all ophthalmologists that a pathological investigation of surgically removed tumor tissue is indispensable for a complete diagnosis and may decisively influence the subsequent therapy. Since tumors always play a special role for the patient, some background on the diagnostic approach by the pathologist is presented: Typing of tumors, thus, their “name” – the diagnostic designation, is based on their cell or tissue of origin regarding ontogenetic derivations. Grading of tumors describes their biological potential and behavior and includes prognosis, thus, the survival time and rate, probability of relapse and durability of remission. Staging of tumors determines their spreading, for instance by invading neighboring structures (e.g. sclera invasion by melanoma). The relevant classification source for staging of tumors is the TNM system (Tumor, Node, Metastasis). Prediction, thus, the probability of response to a specific therapeutic agent or modality, is based on molecular testing by means of predictive markers. Molecular properties of a tumor or molecular differences within a single tumor entity are identified being decisive for a desirable response to a specific therapy [1].
Ophthalmologists may be less aware of that pathology can contribute to ensure the diagnosis in a broad range of non-tumorous eye diseases by use of various methods such as conventional histological staining, enzyme-histochemistry, immunohistochemistry, immunofluorescence, molecular pathology, in-situ hybridization, ultrastructural methods, such as transmission and scanning electron microscopy, and more and more methylation profiling, the latter a recent method to improve tumor classification. Thus, in non-tumorous cases too, clinical neuro-ophthalmopathology serves as decisive support for ophthalmologists by confirming the clinical diagnosis or establishing a hitherto unsure diagnosis. In appropriate cases by use of a broad range of methods available in neuropathology, it is possible to provide information on heredity, prognosis and prediction of therapeutic success or failure.
Since there is an anatomic overlapping in different cases of eye diseases with extension of the pathology into neighboring structures or even with a systemic character, some diseases of the eye must be treated in close co-operation, including transdisciplinary research, between clinical neuroscientists such as ophthalmologists, neurologists, neurosurgeons and specialized neuropathologists/pathologists. Thus, tumorous and non-tumorous exemplary cases illustrate that, on the one hand, the pathology of the eye may help to diagnose various syndromes affecting the nervous system and beyond, and, on the other hand, a pathological investigation remote from the eye – such as a muscle biopsy of the thigh in mitochondrial diseases – may be helpful to establish the final ophthalmological diagnosis.
For detailed questions standard handbooks of ophthalmopathology should be consulted such as the first pioneer handbook of pathology of the eye by Naumann [2,3], the WHO International Classification of Tumours of the Eye and its Adnexa [4] or the most recent AFIP Atlas on Tumors of the Eye and Ocular Adnexa [5]. However, ophthalmopathology is more than just tumors.
For the present review, the morphology of some frequent and rare, yet typical exemplary cases of neuro-ophthalmopathology are compiled including tumors and non-tumorous cases as well as systemic diseases and syndromes in which histopathology can contribute to establish the correct ophthalmological diagnosis.
Results
Pathology of the Eyelid
The chosen examples are tumors of the eyelid.
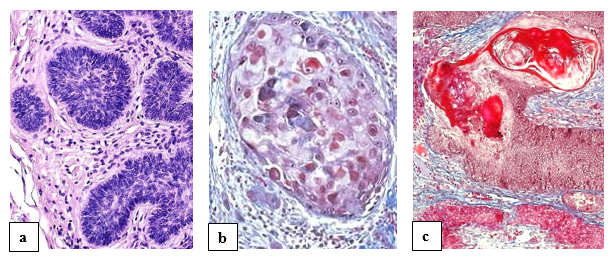
Basal cell carcinoma (BCC) or synonymously basalioma is the most frequently diagnosed type of skin cancer, with eyelid accounting for a notable proportion (figure 1a). The most common histologic subtypes are infiltrative, nodular, and basosquamous basal cell carcinomas. It is of utmost importance for the pathologist to ensure histologically tumor-free resection margins. Adverse prognostic factors associated are previous recurrences, aggressive histologic subtypes, longer duration of lesion, larger lesion size, and the presence of perineural invasion. Orbital invasion is possible [6].
Squamous cell carcinoma (SCC) of the eyelid or synonymously spinalioma [7] behaves in the same malignant way as SCCs of the skin elsewhere (figure 1b). The molecular genetics of BCC and SCC, also in differential diagnosis with sebaceous tumors including sebaceous carcinoma, Merkel cell carcinoma and melanoma, are reviewed by Milman and McCormick [8].
Benign follicular tumors comprise a large and heterogeneous group of neoplasms that share a common histogenesis and display morphological features resembling one or several portions of the normal hair follicle. An example for a benign tumor is pilomatrixoma, synonymously pilomatricoma or calcifying epithelioma of Malherbe, which is commonly found in the head and neck region including the region of the eyelid (figure 1c). It is the most common dermal hair follicle tumor and usually presents as an isolated lesion rarely undergoing malignant transformation. It is not uncommon for these tumors to be misdiagnosed [9].
Pathology of the Cornea
The chosen examples are corneal dystrophies with distinct histopathology.
Corneal dystrophies (figure 2) are autosomal dominant disorders of the corneal stroma [10-12]. They are bilateral, progressive conditions characterized by the formation of opacities arising due to insoluble deposits in the corneal stroma leading to visual impairment.
Corneal dystrophies typically result in a gradual bilateral loss of vision in a primary 'white eye' often in conjunction with epithelial defects in later stages. Different forms exist which need appropriate diagnostic approaches, among them granular corneal dystrophy (GCD), Avellino corneal dystrophy (ACD), lattice corneal dystrophy (LCD), and Reis-Bücklers' corneal dystrophy (RBCD).

Ocular Trauma
The chosen examples are intraocular manifestations of blunt or penetrating trauma.
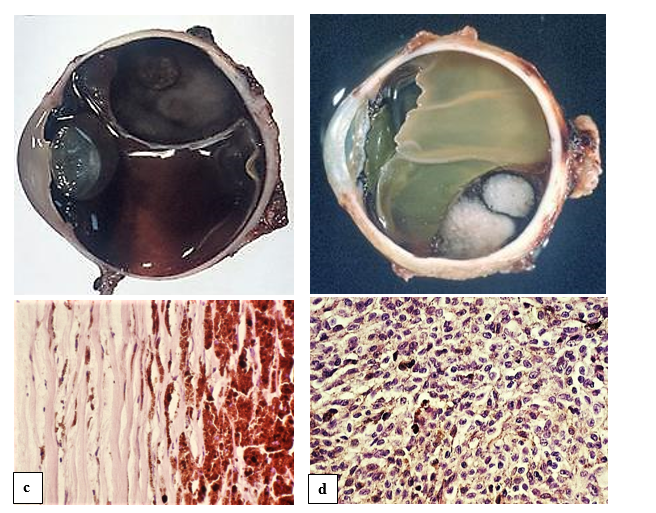
In blunt trauma (figure 3a and b), especially in cases with scleral rupture, intraocular hemorrhages usually necessitate the enucleation of the bulb [13]. In older not treated trauma, organization of the bleeding in the way of calcification can occur (figure 3c).

In penetrating trauma, the main complication – also as undesirable complication of ophthalmosurgical perforating operation – is bacterial infection. Possibly leading to loss of the eye by purulent inflammation (figure 3d). The importance of multidrug-resistant bacterial strains and their role in etiology of inflammatory eye diseases has increased [14].
Inflammatory Diseases
The chosen examples are special forms of retinitis and vasculitis.
Cytomegaly virus (CMV) infection is a common complication in patients with immune depression such as in HIV/AIDS. CMV retinitis is characterized by numerous granular exudative spots in the retina with retinal breaks, retinal necrosis and detachment (figure 4a). CMV infection can also be confirmed in vitreous sample positive for cytomegalovirus DNA by PCR.
Toxoplasmosis, most of the time harmless, may cause in immunocompromised hosts toxoplasmic chorioretinitis and retinal necrosis (figure 4b). In histopathology, the cysts containing the small protozoan parasites confirm the diagnosis [15].

Ophthalmologic manifestations of systemic vasculitis are quite frequent [16]. Granulomatosis with polyangiitis (GPA), synonymously known as Wegener's granulomatosis, is a primary system necrotizing vasculitis of small and medium-sized vessels which is characterized by foci of granulomatous inflammation with multinucleated giant cells (figure 4c). One of the target organs in GPA is the eye. The affection varies in severity, in single cases developing to complete blindness, including bilateral one. GPA can affect any portion of the eye. However, the retina involvement is rare but occurs as sporadic cases in the literature [17]. GPA retinitis may be interpreted as focal retinal inflammatory vasculitis. An obstruction of the central artery of the retina may occur in Wegener's granulomatosis as well.
Another common vasculitis directly involving the eye, in single cases leading to visual loss and to be differentiated from GPA, is giant cell arteritis (GCA), synonymously Horton’s arteritis. GCA is a systemic immune-mediated vasculitis [18] not directly involving the retina but has a predilection for the extradural cranial arteries which include the ophthalmic and the posterior ciliary arteries. The chronic granulomatous inflammation affects the walls of large and medium-sized arteries (figure 5).

Temporal artery biopsy remains the gold standard for diagnosis. However, inflammation can involve several ocular structures, including the sclera, retina, uvea, and cause vascular changes in these tissues. Retinal vasculitis is the most common finding associated with uveitis.
Intraocular Tumors
The chosen examples are retinoblastoma, typical for childhood, and melanoma, the most frequent intraocular tumor.
Retinoblastoma (figure 6) is an autosomal dominant rare childhood tumor of the developing retina initiated through the biallelic inactivation of the retinoblastoma tumor susceptibility gene (RB1). MDM2 and MDM4 genes may also play an important role in retinoblastoma tumorigenesis [19].
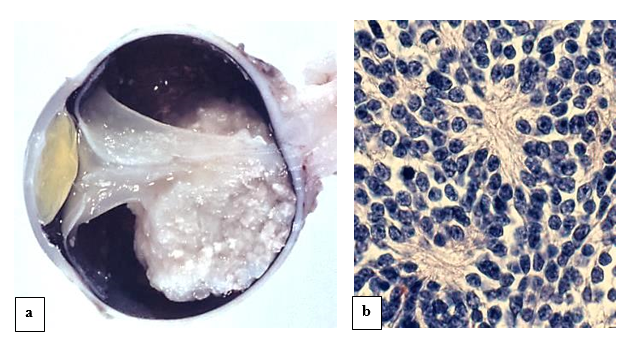
Fine needle aspiration cytology may be a useful diagnostic modality [20]. However, this diagnostic modality of retinoblastoma is invasive and may lead to the spread of tumor cells. Due to this reason, better ways of diagnosis are being explored. Studies on the exosomes in tissue and serum might help designing better diagnostic approaches for retinoblastoma. Exosomes have been characterized as nanometer-sized extracellular double-membrane lipid bilayer bio-vesicles secreted by the fusion of vesicular bodies with the plasma membrane. Tear fluid consists of exosomes that are shown to regulate various cellular processes [21].
Melanoma (figure 7) is the most common intraocular tumor and affects the uvea including the pigmented cells of the iris, muscle fibers of the ciliary body, and the choroid layer of blood vessels.In addition to histopathology, uveal melanoma genetics for assessing prognosis and predicting response to targeted molecular therapy has become indispensable. More recently, gene expression profiling has provided a highly accurate and biologically informative gold standard for molecular prognostication.
Molecular testing of major driver mutations in melanomas that are associated with different growth behavior and prognosis, allows the prediction of response to targeted molecular therapies. Mutations in GNAQ and GNA11 are early events that promote cell proliferation. Mutations in BAP1, SF3B1, and EIF1AX are later events that are to a large extent mutually exclusive. Mutations in BAP1 are strongly associated with metastasis, whereas those in SF3B1 and EIF1AX are associated with good prognosis [22].
Tumors and Tumor-like Lesions in the Orbit
The most common tumors in the orbit, i.e. pilocytic astrocytoma of the optic nerve, meningioma of the meninges of the optic nerve, but also tumors like lymphoma or benign lipoma behave as in other parts of the body. The chosen examples are non-tumorous tumor-like lesions, namely cavernous hemangioma and dermoid cyst.
Orbital cavernous hemangioma (figure 8) is a rare vascular malformation which may become clinically apparent by pulsatile proptosis or slow progressive diplopia. Histologically, densely packed thin-walled blood vessels of different size occur. The hypercellularity and stromal changes identified can be understood within the pathogenic context of thrombosis and recanalization in the lesion [23].

Dermoid cysts (figure 9) can occur in the orbital region as benign congenital teratoid tumors which can be regarded as choristomas, thus, mimicking all skin layers, i.e. epidermis, dermis and subcutaneous fat and, therefore, contain squamous keratin, fatty substances from sebaceous glands and even hairs originating from included hair follicles. Superficial orbital dermoid cysts are more frequent and present earlier in life [24]. In adults, deep orbital dermoid cysts are more likely to present with frontal bone erosion extending into the frontal sinus [25]. An inflammatory reaction can occur if a dermoid cyst is disrupted.

Phakomatoses
The chosen examples are tuberous sclerosis complex (TSC) and Von Hippel-Lindau disease (VHL).
Phakomatoses, are hereditary multisystem disorders whereby mutation of tumor suppressor genes causes hamartomas which carry the risk of malignant transformation. Ophthalmological features found in phakomatoses were recently reviewed [26,27]. Lisch and choroidal nodules in neurofibromatosis type 1, retinal astrocytomas in tuberous sclerosis complex, and retinal capillary hemangioma in Von Hippel-Lindau disease are the principal ophthalmic hamartomatous manifestations.
Tuberous sclerosis Bourneville-Pringle or tuberous sclerosis complex is characterized by hamartomas of the heart, kidney, brain (subependymal giant cell astrocytoma), skin (adenoma sebaceum), and eyes. Retinal astrocytomas or hamartomas are the most common ocular finding in tuberous sclerosis (figure 10a). Most hamartomas are non-progressive. Again, not only clinical features and ocular findings, but also histopathology and genetics play a crucial role for the correct diagnosis of TSC [28].

Retinocerebellar angiomatosis Von Hippel-Lindau is an inherited, multisystemic phakomatosis involving retina (figure 10b) and cerebellum (capillary hemangioblastoma). Affected persons may also have angiomatous or cystic lesions of the kidneys, pancreas, and epididymis, as well as adrenal pheochromocytomas and clear cell type renal cell carcinomas. The VHL gene as responsible tumor suppressor gene was also found to be responsible for tumorigenesis in the corresponding sporadic tumors, especially in the clear cell type of renal cell carcinomas. VHL can be classified in type 1 (without pheochromocytoma) and type 2 (with pheochromocytoma). Type 2 is further classified as type 2A (pheochromocytoma is present along with CNS hemangioblastomas but no RCC), type 2B (pheochromocytoma is present along with both CNS hemangioblastomas and RCC), and type 2C (pheochromocytoma is present without hemangioblastomas or RCC) [29].
Malformations
The chosen example is cyclopia.
Cyclopia is a rare and lethal complex human malformation, resulting from incomplete cleavage of prosencephalon into right and left hemispheres, the holoprosencephaly (figure 11a), occurring between the 18th and the 28th day of gestation [30]. Cyclopia typically presents with one median eye with a single iris, the ‘true’ cyclops, or two fused eye globes in a single orbit, the synophthalmic cyclopia (figure 11b).

Instead of the absent nose a proboscis (nose anlage with an incomplete induction of nasal structures) is situated above the eye. Mutations in the four main genes involved in holoprosencephaly (SHH, ZIC2, SIX3, TGIF) were identified in 25% of cases [31,32]. Somatic mosaicism in developmental gene mutations should be considered a causal factor for variable phenotypes, sporadic cases, and de novo mutations in human developmental disorders [33].
Mitochondrial Disorders
Mitochondria are found in all nucleated human cells to meet the needs of various tissues, particularly those with high energy demand such as nervous cells or muscle tissue. In the case of mtDNA, the rule is “maternal inheritance”, thus, paternal mtDNA from the sperm is eliminated and only maternal mtDNA from the oocyte is transmitted to the offspring.
Mutations in mtDNA-genes or in mitochondria-related nDNA-genes can result in mitochondrial dysfunction [34]. To date, more than 1,700 mitochondrial genes are known, of which more than 300 are associated with mitochondrial diseases.
The principal histological hallmarks of mitochondrial myopathy and encephalopathy, often associated with ocular features, are ragged red fibers (figure 12) in skeletal muscle, including extraocular muscles, as common finding determining the diagnosis of mitochondriopathy. Single muscle fibers are ‘ragged’ and, in suitable staining, appear red from abundant mitochondria. Therefore, a muscle biopsy may be helpful, especially if specialized genetic investigation and counseling are not available. In a variety of mitochondriopathies, common central nervous system findings are seizures, dementia, migraine, stroke-like episodes, ataxia, and spasticity [35].
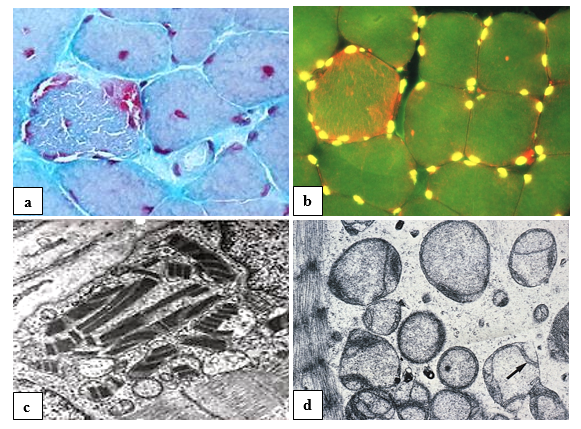
Mitochondrial disorders are a clinically heterogeneous and may occur at any age, some disorders only affecting a single organ (e.g., the eye in Leber hereditary optic neuropathy [LHON]), others involving multiple organ systems and often presenting with prominent neurologic and myopathic features. Typical syndromes comprise the Kearns-Sayre syndrome (KSS), chronic progressive external ophthalmoplegia (CPEO), mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS), myoclonic epilepsy with ragged-red fibers (MERRF), neurogenic weakness with ataxia and retinitis pigmentosa (NARP), Leigh syndrome (LS) or the mitochondrial recessive ataxia syndrome (MIRAS) [36,37].
However, considerable clinical variability exists. For instance, in Kearns-Sayre syndrome, systemic involvement includes CPEO, with ptosis being the most common complaint, and cardiomyopathy. Other variable features are short stature; cerebellar symptoms; weakness of muscles of the face, pharynx, trunk, or extremities; and progressive hearing loss [38].
Neuro-ophthalmopathology is indispensable for surgical ophthalmology, often in co-operation with neurosurgeons, improving surgical effectivity, quality control of results and diagnostics. For correlation and evaluation of morphological findings in daily ophthalmological diagnostics from slit lamp to OCT, histopathological findings, electron microscopy and a variety of other pathological methods can add important information for the understanding of pathogenesis and evolution of ocular diseases.
However, certain ocular diseases as well as systemic syndromes also affecting the eye can exactly be diagnosed by means of genetic investigations only.
Be aware that neuro-ophthalmopathology is not restricted to the anatomic area of the eye, thus, also neuropathology and myopathology may provide important information for a comprehensive ophthalmological diagnosis.
Statement of Ethics
Ethical approval was not required for this study in accordance with national guidelines since no individual patients are shown.
Disclosure Statement
The author has no relevant financial or non-financial interests to disclose. The author has no conflicts of interest to declare that are relevant to the content of this article.
Funding Sources statement
This study did not receive any funding or grant support.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
