
Review Article | DOI: https://doi.org/10.31579/2692-9562/132
Department of Pharmacy, University of Karachi, Pakistan.
*Corresponding Author: Rehan Haider, Department of Pharmacy, University of Karachi, Pakistan.
Citation: : Rehan Haider, Asghar Mehdi, Geetha Kumari Das, Zameer Ahmed, Sambreen Zameer, (2024), Quinolones: Development, Mechanism of Action, and Clinical Applications of Synthetic Antibiotics, Journal of Clinical Otorhinolaryngology, 6(5); DOI:10.31579/2692-9562/132
Copyright: © 2024, Rehan Haider. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited
Received: 09 September 2024 | Accepted: 25 September 2024 | Published: 30 September 2024
Keywords: quinolones; antibiotics; synthesis; antibacterial activity; DNA gyrase; topoisomerase IV; drug development; antibiotic resistance; chemical structure; pharmacokinetics
Quinolones, a class of synthetic antibiotics, have garnered significant attention for his or her broad-spectrum antibacterial activity and various therapeutic application. This paper provides a comprehensive review of the synthesis and antibacterial properties of quinolones, emphasizing their chemical structures, mechanisms of action, and current advancements in drug development. The synthesis of quinolones involves complex chemical transformation, with ongoing research aimed at enhancing their efficacy and yield. Structural modification, particularly substitutions at key positions, have led to the development of quinolone derivatives with improved pharmacokinetic properties and enhanced antibacterial potency. This overview also discusses the challenges and future direction in the development of quinolone antibiotics.
Quinolones are a prominent class of synthetic antibiotics broadly used to treat numerous bacterial infections. since that their discovery, quinolones have evolved extensively, with numerous structural adjustments enhancing their efficacy and broadening their spectrum of the activity. This paper goals to provide a detailed review of the synthesis strategies, antibacterial mechanisms, and latest-advancements in the field of quinolone antibiotics. Understanding this aspect is critical for the ongoing improvement and optimization of quinolone-based therapies.
After the idea of discriminating toxicity in chemotherapy was brought in at first, In the 20th centuries (Ehrlich, 1913),{1} classes of substances with decontaminating characteristics, produced by microorganisms or forged through combination, were obtained. After the finding of penicillin, the first medicine made acquainted in dispassionate use in man in the 1940s, various types of medicines were produced. Antibiotics to a degree tested—β-lactams, macrolides, aminoglycosides, and tetracyclines—were found and introduced, all with an intensely short ending. These were obtained either by seclusion from fungi or by chemical qualification of the normally isolated substrates. These ruled the antimicrobial manufacturing, while artificial means obtained Essences play only a theatrical role. (Chu & Fernandes, 1991) {2}
In 1962, G. Y. Lesher and his collaborators {3} imported the first quinolone derivative, nalidixic acid (1-ethyl-1,4-dihydro-7-methyl-4-oxo-1,8-naphthyridine-3-carboxylic acid), (1, Lesher and others. (1962) that had moderate activity against grandmother-negative structures and was secondhand to treat urinary contamination. In the following age, a large gamma of descendants of the prevailing pieces was synthesized, which may have been gathered by (cinoxacin), pyrido-pyrimidine (pipemidic acid; piromidic acid), naphthyridine (nalidixic acid), and quinolones (oxolinic acid, miloxacin, tioxacin, etc.). These derivatives,accompanying changed forms, have two universal pharmacological properties that admitted the ruling class to be the top secret as the first production of biologically active derivatives accompanying quinolone construction.
The two prevalent characteristics of first-generation quinolones are as follows:
A narrow, completely clean range, designed exceptionally for Enterobacteriaceae - a pharmacokinetic that allows for rapid removal and weakened fabric absorption, admitting bureaucracy expected secondhand as a urinary antiseptic.
Structural Variation Quinolones
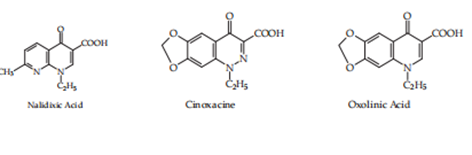
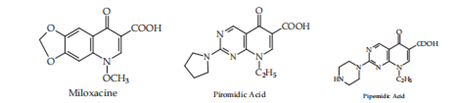
Figure. 1. First-generation quinolones
The benefit of the first creation of quinolones spurred the research circumference, which led to the obtaining or receiving, through combination, afterwards 1980, of a new series of compounds accompanying more forceful antibacterial features and a fuller spectrum of decontaminating ventures that included Gram positive, certain and gram-negative animals, and that place defined by their strength expected applied to all the local contaminants. Koga and his co-workers imported Norfloxacin into dispassionate use in 1980, the first quinolone with a fluorine bit substituted at the C-6 position and a piperazine C-7. Norfloxacin (Koga and others. 1980){4} was the first quinolone with raised antimicrobial exercise, pursuing a large range of grandam positive and gram negative microorganisms including Pseudomonas aeruginosa
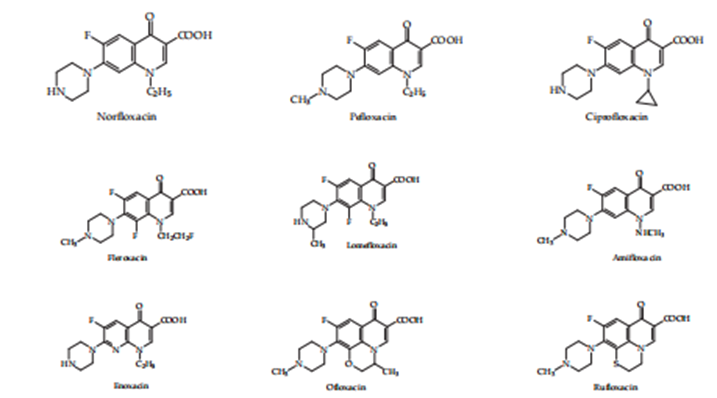
Figure 2. Second-generation quinolones.
The research on derivatives accompanying quinolone construction has led to new compounds acquired currently, which have existed top-secret as triennial and fourth creation intrinsic quinolones, which are largely effective against Staphylococcus aureus. Their big, completely clean spectrum includes anaerobes, Chlamydia, and Mycoplasma. (Brighty & Gootz, 2000) {5}
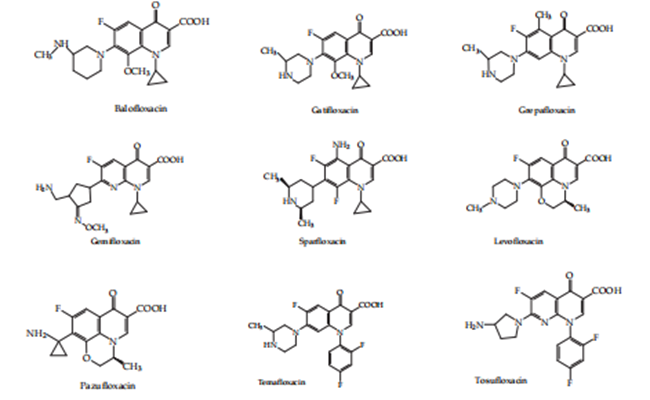
Figure 3: Third- generation quinolones.
The four productions have the following coarse aspects: a similar method of action by restriction, A subunit of DNA-gyrase, a particularly chromosomal bacterial opposition and a few similar microorganisms: photo toxicity, neurotoxicity, and animate skeleton toxicity.
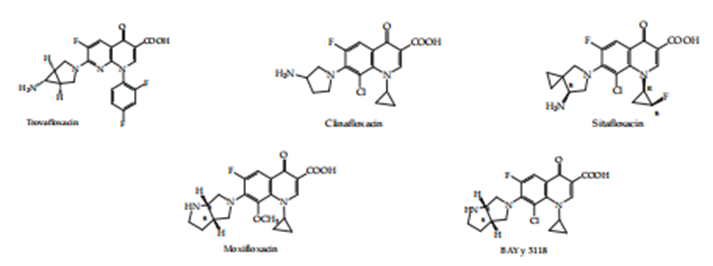
Figure. 4. Fourth- generation quinolones.
Until now, a lot of antibacterial essences owned by the above-noticed This class happened used in medicine. Quinolones are secondhand when considering infections of the urinary area, the respiring tract, stomach contaminations, ear/nose/neck contaminations, STDs, soft fabric and skin infection, meningitis caused by Gram-negative bacteria, and Staphylococci microorganisms, liver and hostility infections, infection of blood and endocarditis, precautions, and surgical infections and cases with immune imperfections. The system of operation of quinolone-uncontaminated powers includes the restriction of DNA gyrase (bacterial topoisomerase II), resulting in a brisk and uncontaminated effect. The uncontaminated exercise of quinolones (calculated in agreements of MIC) is still the result of consolidation of bacterial container seepage and DNA gyrase inhibitory activity. The decontaminating project of quinolones depends not only on the bi cyclic hetero aromatic method joining the 1,4-pyrido-4-pyridine-3-carboxylic acid subdivision and a pungent ring but also on the character of the minor substituents and their relation to space relationships. These substituents utilize their influence on a bacterial project by providing supplementary closeness for Bacterial enzymes, embellishing container seepage, or changes in pharmacokinetics. In general, research on ideal quinolones persists. Such a quinolone must be biologically alive on a large range of grandam definite and grandam negative microorganisms, aerobes and anaerobes and mycobacteria must have as few aftereffects as likely, which is wonderful solubility in water, and spoken bioavailability. In Figure 5, the ultimate common combined synthetic differences got all the attention of the research for new quinolones accompanying decontamination ventures.
1. Quinolones: Structural countenance and procedure of synthesis
2. 2.1 Structural face Quinolone products are a major class of uncontaminated power that accompany expansive operations. Basic construction of these compounds (Figure 6) is a bi cyclic construction, which holds a ring of type A 4-piridinona linked accompanying savory or hetero aromatic ring B. The ring aggressive personality 4-Piridinona is a ring accompanying certain needs: an unsaturation available in 2–3, a free acid function prepared 3, and a substituent at the nitrogen at position 1.
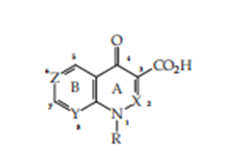
Figure 5: Structural variations of the most recent quinolones.
2.1.1 Bi cyclic quinolones
Position 1
The studies on quinolones indicated that for the compound to have a decontaminating venture, the N-1 position requires a substituent. Many quinolones are present in the N position. ethyl (norfloxacin, pefloxacin, and lomefloxacin), fluoroethyl (fleroxacin), vinyl, chloroethyl, trifluoromethyl, aminoethyl, cyclopropyl (ciprofloxacin), t-butyl, bi-cyclopentyl, p-fluorophenyl, 2,4-fluorophenyl (Scott 1997) {6}
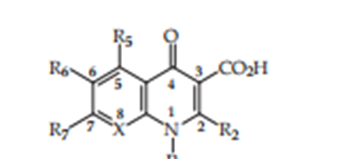
Figure. 7. Basic structure of bi cyclic quinolones.

R = H, methyl, ethyl, substituted phenyl
R6= F, Cl
R7= hetero cycle
Figure 8,7: Substituted-6-halo-4-oxo-4H-[1,3]-thiazeto[3,2]quinolin-3-carboxilyc acid.
Position 2
Quinolones were held at the C-2 hydrogen (R2=H). The substitute for hydrogen has mainly demonstrates the expected disadvantages. However, few compounds hold an appropriate C-1, C-2 The ring is currently used to seize organic exercises. (Figure 7, Segawa 1992) {7} (Figure 8 (Scott, 1997) C-3 carboxylic acid subdivision is most often confronted. (Chu & Fernandes 1991). In the late 1980s, a qualification was depicted that removed the need for C-3 carboxylic acid. A melted isothiazolinone ring was labeled as a symbol of a carboxylic acid mimic. Compound A-62824 (Figure 9). have been found with biological activity

Figure. 9. Quinolones with sulfur substituent at C-2.

Figure. 10. A-62824.
Position 4
The C-4 oxo group of the quinolone core is mainly deliberate and expected to be essential for uncontaminated venture. Position 5
The choice of the C-5 substituent performs expected commands for one of the steric rules and the types of N-1 and C-8 substituents (Chu & Fernandes, 1991). (R5 = methyl, element, amino) when X = CF)
Position 6
The type of C-6 substituent has an excellent effect on the DNA-gyrase inhibitory exercise and container seepage. (Domagala et al., 1986) {8}. The R6 maybe H, Cl, F, NO2, NH2, CN, CH3SCH3, COCH3 (Koga and others, 1980)
Position 7
The choice of the C-7 substituent is a key issue that persists in guiding the design of new Uncontaminated Quinolones. R7 can be substituted with or unsubstituted piperazine. amino pyrrolidines and aminoalkyl pyrrolidines (Figure 5) (Chu and Fernandes 1991) (Scott 1997).
Position 8
The most accepted alternatives at the C-8 position are hydrogen modicum (X= CH) or a nitrogen Jot (naphthyridine) (X=N). However, the compact lipophilic group (X = CF, C-CF3, CCl, and C OCH3) increases the quality of the venture. (Chu and Fernandes, 1991; Scott, 1997).
2.2 Method of synthesis
Gould-Jacob’s pattern
Quinolones are artificial in origin. (Chu & Fernandes, 1991). The lowest artificial method to make quinolone descendants is the Gould-Jacobs procedure (Figure 11). This order is secondhand primarily for a combination of compounds accompanying

Figure 11. Gould-Jacob’s method.
N-1 alkyl substituents and resides in the abridgment of anilines (II) accompanying diethyl ethoxymethylenemalonate (EMME), and the cyclization of anilinomethylene malonate. Thermal cyclization can be carried out in dow term (Koga, others, 1980), chlorosulfonic acid, oleum acid, or a combination of chlorosulfonic acid, and oleum acid (Saukaita and Gupton, 1996). The key go-between acquired (IV or X) will sustain an alkylation (Koga and others, 1980), cyclo alkylation accompanying bromo cyclopropane (Kazimierozaack & Pyznar, 1987) {9}, (Sanjose & Ulpiano, 1986) {10}, or accompanying 1-Bromo-1-ethoxy-cyclopropane, arylation accompanying para-nitro-chlorogenic or 2,4-dinitro-chlorogenic (Raddl &Zikan, 1989) {11} in consideration of placing the substituent prepared 1 of the quinolone cores. The ethyl ester (V) sustains a hydrolysis response, and the quinoline-3-carboxylic acid (VI), following regiospecific replacement of the 7-chloro group leads to the ending compounds (VIII). Figure 11 exemplifies again, orders for combining 1-cyclopropyl-quinolones offset for the anilines in recipe II. Anilines (II) is reacted accompanying 1-bromo-1-ethoxy-cyclopropane (Ramos & Garcia, 1994; Scriewer and others, 1988) {12,13} or a cyclopropyl-metallic compound (McGuirk 1989) {14}. Alternatively, N-ethyl substituted anilines of rule XV concede the possibility of being made by diminutive amination accompanying an appropriate aldehyde and an appropriate lowering

Figure 12: The modified Gould-Jacobs method.

Figure. 13. Method requiers the reaction of isatoic anhydride with sodium ethyl formyl acetate
Power: diborane, palladium on element accompanying hydrogen, sodium borohydride or sodium cyano borohydride (McGuirk) 1989) (Ramos 1994). N-isopropyl-substituted quinolones can be formed by alkylation accompanying Isopropyl bromide (Pintilie and others, September 2009) {15} (Pintilie et al., Oct. 2009),{16} (Pintilie and others, 2010) {17}, of compounds IV. (Figure 11). The changed Gould-Jacobs procedure will more be secondhand, placing the diethyl Ethoxymethylenemalonate reacted with mono substituted N-aniline (XVII) (Figure 12). The aniline (XVII) is obtained by subtractive amination of ketones and aldehydes accompanying sodium borohydride-tart acid (Itoh & Kato 1984) {18} or triacetoxyborohydride. (Pintilie et al. 2009). (Pintilie and others. 2010){19}
The method requires the backlash of isotonic anhydride with sodium ethyl formyl acetate Another combination order requires the response of isotonic anhydride accompanying sodio-ethyl formyl acetate (Figure 13). 2,4,5-trihalobenzoic acid (XX) is reacted with an appropriate amine, and before is acted upon, accompanying the compound: R2R3CO (R2 = R3 = Cl, CCl3O or R2= C1-10 alkyl, and R3 = Cl) to produce benzoxazindione (XXII). Therefore, the benzoxazine dione (XXII) was shortened to the accompanying compound (HOCH=CHCO2Et) to specify the key compound (V). Intramolecular nucleophilic dislocation cyclization route to quinolones (a) An effective and regiospecific combining by way of an intramolecular nucleophilic dislocation
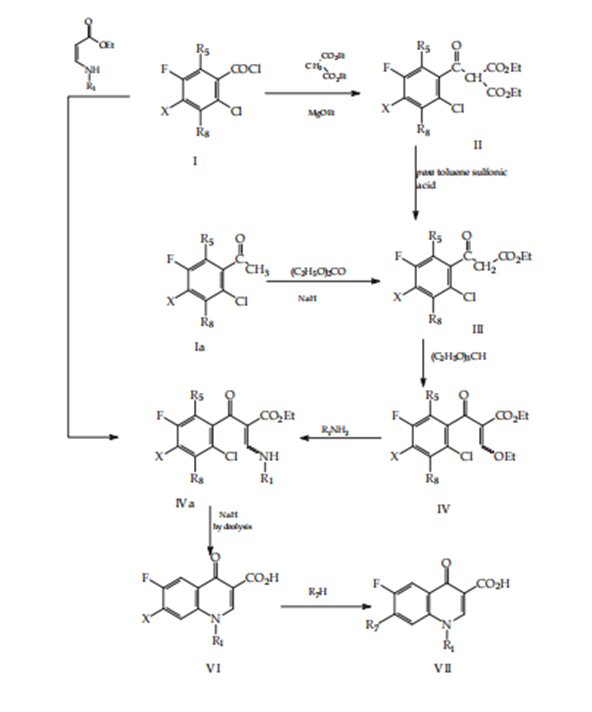
Figure. 14. Intramolecular nucleophilic displacement cyclization route to quinolones (a).
The cyclization response is also described. (Chu, 1985) (Figure 14). The key compound (III) may be acquired by
• backlash of benzoic acid chloride (I) accompanying ethyl malonic acid; the compound (II) present the compound (III).(Petersen & Grohe 1984a),{20} (Petersen & Grohe 1984b),{21}
• acetophenone (Ia) is concentrated along with diethyl carbonate in the demeanor of sodium hydride (Chu and others, 1985){22} Intermediates (III) react with accompanying tart anhydride in the ghost trietilortoformiate to produces 3-ethoxy-2-benzoyl-ethyl acrylate (IV). Compound (IV) is further reacted to, accompanying an appropriate amine in dichloromethane at range hotness to specify 3-anilino-2-benzoyl ethyl acrylate (IVa). Compound (IVa) may still be derived from benzoic acid chloride (I). (Chu & Fernandes 1991). Treatment with a base induces cyclization, producing quinolones (V). Intramolecular nucleophilic dislocation cyclization route to quinolones (b) A combination order similar to that characterized above is shown in Figure 15. This design includes intramolecular cyclization of compound (VIII). (Egawa and others. 1987).{23}
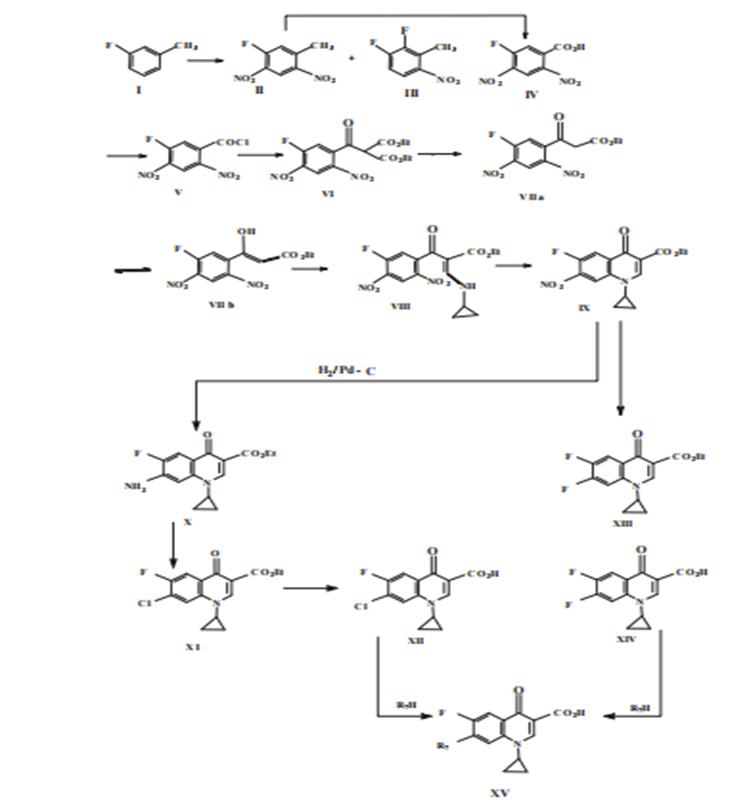
Figure. 15. Intramolecular nucleophilic displacement cyclization route to quinolones (b).
2.3 Structure of the New Compounds
This paper presents an exploratory dossier concerning the combination of various quinolones accompanying approximate rule: (Figure 16)
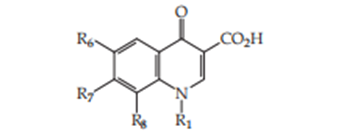
Figure. 16. The structure of the new compounds.
R1 = ethyl, isopropyl, 2-buthyl, 2-penthyl, benzyl, alkyl, p-nitrophenyl, p-amino-phenyl
R6 = hydrogen, fluor, chlor, methyl
R7 = 3-methyl-piperazinyl, 4-methyl-piperazinyl, piperidinyl, 3-methyl-piperidinyl,
4-methyl-piperidinyl, pyrrolidinyl, morpholinyl, and homopiperazinyl
R8 = hydrogen, chlorine, methyl, methoxy, nitro
2.4 Synthesis Pathway
The combination of the novel quinolones underwent a Gould-Jacobs cyclization process (Figure 17). An appropriate unsubstituted aniline (1) is reacted with accompanying diethyl ethoxy methylene malonate (EMME) to produce anilinomethylenemalonate (2). An after-warm process induces Gould-Jacob’s cyclization to afford the corresponding 4-hydroxyquinoline-3-carboxylate ester (3). (R6 = fluoro, chloro, methyl, and hydrogen) (Pintilie and others.2009a) (Pintilie et al. 2009b) (Pintilie et al. 2010).

Figure. 17. Synthesis of the new quinolones.
The following step is the alkylation of the 4-hidroxy-quinoline-3-carboxylate ester (3). that is customarily skilled by responding with an appropriate alkyl halide, dialkyl sulfates, and covering halide to produce quinolone 3-carboxylate ester (6). (R1 = ethyl, allyl, benzyl, and p-nitrophenyl)
(Pintilie and others, 2003a); (Pintilie and others, 2003b); (Pintilie and others, 2003c);{24} (Pintilie & Nita, 2011). A reduced approach resorts to the use of a mono substituted aniline (4) as an offset material to avoid N-1-amine alkylation (R1 = isopropyl, 2-butyl, 2-pentyl). (Pintilie and others, 2009a); (Pintilie and others, 2009b); (Pintilie and others, 2010). A powerful acid (to a degree, polyphosphoric) acid) is frequently used to encourage cyclization, which straightforwardly occurs in the composition of N-isopropyl-4-oxo-quinolone-3-carboxylate ester (6) (R1 = isopropyl, 2-butyl, 2-pentyl). In either case, the ending guidance is acid or fundamental hydrolysis to split the ester and create the biological Five free carboxylic acids (7). The biologically
alive free carboxylic acid (7) was further acquired from the equivalent 4-hidroxy-quinoline-3-carboxylate ester (3) by alkylation accompanying dialkyl sulfates in the presence of the opposite of an acid; for instance, the response it can skillfully complete activity in liquid 40% sodium hydroxide resolution. The dislocation of the 7-chloro group was accompanied by hetero cycle-allowed compounds (8). The combining of new 1-aryl quinoline-3-carboxylic acids corresponds to Figure 18. compound (3) (R6=F, Cl, CH3) is a direct N-arylation reaction. Treatment of (3) accompanying potassium carbonate in DMSO and p-fluoro-nitrobenzene (9). The esters were hydrolyzed to the appropriate acids (10) by refluxing, accompanied by a combination of hydrochloric and tartaric acids, upon the situation accompanying a hetero cycle surrendered compound (11). 1-(p-Aminophenyl)-quinoline-3-carboxylic acid (12) may be caused by a prevalent decline in the nitro group using sodium dithionite.

Figure. 18. Sinthesys of 1-aryl-quinolones.
The synthesis of the new 8-substituted quinoline-3-carboxylic acid is shown in Fig. 19. 8-Chloro-quinoline-3-carboxylic acids (13) were combined from 8-unsubstituted quinoline-3- carboxylic acids by chlorination with sulphuryl chloride. 8-Methoxy-quinoline-3-carboxylic Acids (14) were arranged to admit compounds (13) to obtain a soluble metallic alcohol.

Figure. 19. Sinthesys of the new 8-substituted quinoline-3-carboxylic acids.
2.5 New compounds: Structure and antimicrobial endeavor
2.5.1 Structure of the New Compounds
A succession of new 4-oxo-1,4-dihydroquinoline-3-carboxylic acids were combined. (Figure 20)

Figure. 20. Structure of the new compounds.
(Table 1)


Table 1. 4-Oxo- 1,4-dihydro-quinoline-3-carboxylic acids synthesized in this paper.
2.5.2 Antibacterial action of the new compounds
The new compounds were judged for “artificial” action by deciding minimum inhibitory aggregation against of microorganisms Escherichia.
Coli, Staphylococcus. Aureus and Pseudomonas. aeruginosa, by agar something for dunking design (Buiuc 1998) (NCCLS 2003). {25,26} (Table 2).
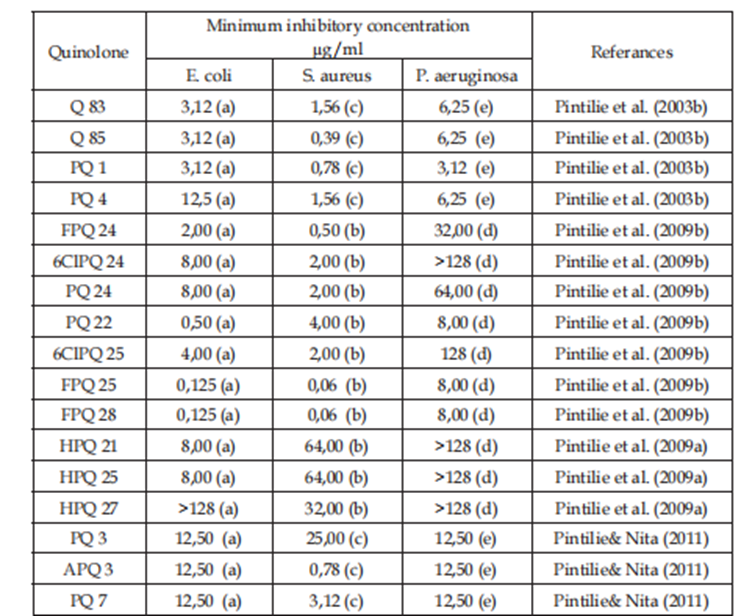
a. Escherichia. coli ATCC 25922, b. Staphylococcus. aureus ATCC29213, c. Staphylococcus. aureus ATCC29223,
d. Pseudomonas .aeruginosa ATCC27813,e. Pseudomonas .aeruginosa ATCC27853
Table 2. “In vitro” antibacterial activity of the new quinolones.
This study uses a versatile approach to investigate the decontamination project of recently synthesized quinolone products. The combining of these descendants was achieved through an order of traditional synthetic reactions, with painstaking concern given to optimizing backlash environments for improved yields. Characterization techniques, including basic attractive resonance (NMR) spectroscopy and bulk spectrometry, were working to validate the structural uprightness of the combined compounds.
Antibacterial venture assessments were conducted using a committee of clinically relevant bacterial strains. Standard microbiological arrangements, containing platter diffusion and minimum inhibitory aggregation (MIC) assays, were working to determine the susceptibility of the bacterial isolates to the quinolone descendants. Quality control measures were executed to guarantee the accuracy and reproducibility of the results
The synthesis of quinolones was carried out using a series of chemical reactions, starting with the cyclization of N-alkylation anthranilic acids. various substituents were introduced at key positions to evaluate their impact on antibacterial activity. The synthesized quinolone derivatives were characterized using NMR, IR, and mass spectrometry.
The introduction of fluorine on the C-6 position and piperazine on the C-7 position resulted in derivatives with significantly enhanced antibacterial activity against Gram- negative bacteria. this modification improved the compounds' lipophilicity and cellular uptake, leading to increased efficacy. The findings suggest that further, structural modifications could yield quinolone derivatives with even broader antibacterial spectra and better pharmacokinetic properties. This study highlights the potential for developing new quinolone antibiotics with improved therapeutic profiles through targeted chemical modifications
In conclusion, we synthesized new quinolones and evaluated their decontaminating capacity. The consequences indicate that substituent combos in the quinolone ring can produce effective decontaminating retailers. particularly, the compound FPQ-28 (1-ethyl-6-fluoro-7-morpholinyl-8-chloro-1,4-dihydro-four-oxo-quinoline-3-carboxylic acid) (determine 21), in step with QSAR research (Tarko et al., 2008), established giant synthetic activity towards E. coli ATCC 25922 (MIC 0. a hundred 25 µg/mL) and S. aureus ATCC 29213 (MIC 0.06 µg/mL).
parent 21. 1-Ethyl-6-fluoro-7-morpholinyl-8-chloro-1,4-dihydro-4-oxo-quinoline-three-carboxylic acid (FPQ-28).
FPQ-28 NMR and IR spectra:
1H-NMR (DMSO-d6, δ ppm, J Hz):
8.97 (s, 1H, H-2)
8.07 (d, 1H, H-5, 11.8)
4.89 (q, 2H, H-17, 7.2)
3.82 (m, 4H, syst. A2B2, H-13-15)
3.37 (m, 4H, syst. A2B2, H-12-16)
1.46 (t, 3H, H-18, 7.2)
13C-NMR (DMSO-d6, δ ppm, J Hz):
175.56 (C-4)
166.12 (C-21)
154.95 (d, J(13C-19F)=254.8, C-6)
158.37 (Cq)
153.04 (C-2)
125.94 (Cq)
124.76 (Cq)
116.86 (Cq)
111.57 (d, J(13C-19F)=23.5, C-5)
98.35 (C-3)
67.23 (C-13-15)
53.64 (C-12-16)
51.58 (C-17)
16.14 (C-18)
ft-IR (strong in ATR, ν cm-1):
3056, 2957, 2895, 2849, 1717, 1615, 1558, 1532, 1492, 1435, 1376, 1300, 1253, 1207, 1102, 1033, 980, 920, 890, 846, 803, 740, 651, 528, 464.
The completion of this research project would not have been possible without the contributions and support of many individuals and organizations. We are deeply grateful to all those who played a role in the success of this project We would also like to thank My Mentor [. Naweed Imam Syed Prof. Department of Cell Biology at the University of Calgary and Dr. Sadaf Ahmed Psychophysiology Lab University of Karachi for their invaluable input and support throughout the research. Their insights and expertise were instrumental in shaping the direction of this project
Declaration of Interest
I at this moment declare that
I have no pecuniary or other personal interest, direct or indirect, in any matter that raises or may raise a conflict with my duties as a manager of my office Management
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Financial support and sponsorship
No Funding was received to assist with the preparation of this manuscript
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
