
Research Article | DOI: https://doi.org/10.31579/2693-7247/098
1 Mother Teresa College of Pharmacy, Hyderabad, Telangana, India.
2 Omega College of Pharmacy, Hyderabad, Telangana, India.
3 Tou PharmaBio Pvt Ltd, Hyderabad, Telangana, India.
*Corresponding Author: Rajakumar Devara
Citation: Rajakumar Devara, Jithan Aukunuru, Mohammed Habibuddin(2022). Preparation, Characterization and Evaluation of Prasugrel Hydrochloride Nanosuspensions: Its enhancement of dissolution rate and oral bioavailability. J Pharmaceutics and Pharmacology Research, 5(8); DOI:10.31579/2693-7247/098
Copyright: © 2022 Rajakumar Devara. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 22 June 2022 | Accepted: 08 July 2022 | Published: 11 July 2022
Keywords: prasugrel hydrochloride, dissolution rate, nanosuspension and bioavailability.
The objective of this study was to investigate nanosuspensions for enhancing the solubility and dissolution rate of Prasugrel HCl (PHCl) so as to reduce the fluctuations in its oral bioavailability. Prasugrel Hydrochloride nanosuspensions were prepared using evaporative precipitation method. After preparation, various nanosuspensions were characterized for particle size, scanning electron microscope (SEM), zeta potential, drug entrapment efficiency (EE), Fourier transform infrared spectroscopy (FTIR), and differential scanning calorimetry (DSC). Solubility and in-vitro release of the drug from nanoparticles are determined. The formulations were characterized using techniques such as powder x-ray diffractometry, scanning electron microscopy, in-vitro dissolution and in-vivo absorption in rats. The dissolution rate and oral absorption of Prasugrel Hydrochloride in the form of nanosuspensions was significantly higher than that of oral suspension and pure drug. All the techniques investigated in this study can be used to enhance dissolution rate and oral absorption of prasugrel hydrochloride and thus can reduce the fluctuations in its oral bioavailability. Nanosuspensions demonstrated to be better and superior technique when compared to other techniques investigated in enhancing oral bioavailability of Prasugrel Hydrochloride. Prasugrel hydrochloride nanosuspension was successfully developed using and the bioavailability is 4 folds increased than its pure drug.
Prasugrel hydrochloride is a novel member of a third generation thienopyridine class of anti-platelet agents1. It is a prodrug. Formulation strategy for the hydrochloride salt of prasugrel is focused on developing an immediate-release tablet for oral administration. A reaction between Prasugrel Hydrochloride and an excipient was observed late in the development stages during manufacture and storage. This reaction leads to a partial and irreversible formation of prasugrel free base in the tablets. Analysing the samples used for clinical phase-3 study indicated that salt-to base formation of at least up to 70% had no clinical impact and a requirement has been included in the finished product specification2. Its base form had significantly lower bioavailability compared to salt form and its bioavailability is tremendously influenced by the changes in the pH3. Because of these reasons, Prasugrel Hydrochloride rather than its base form was selected for final development and marketing. Solubility determination results and permeability and metabolism information suggest that Prasugrel Hydrochloride is a BCS class-II compound4. Dissolution is affected by pH and decreases with increasing pH. The bioavailability of Prasugrel Hydrochloride exceeds 79%. The peak plasma concentration of the active metabolite occurs 30 minutes after dosing. The parent compound is not detectable in the plasma. Although the oral bioavailability is generally high, it could be variable because of its poor solubility5. Its aqueous solubility is 2.37e-03 g/L. At low pH values from pH 1 to pH 4, it is slightly soluble and above this pH value is it is insoluble6. As Prasugrel Hydrochloride is a poorly soluble compound, many problems are encountered in its systemic delivery and achievement of therapeutic action. One of the critical problems associated with poorly soluble drugs is too low bioavailability and/or erratic absorption. The oral bioavailability/erratic absorption depends, on several factors including aqueous solubility, drug permeability, dissolution rate, first pass metabolism. As a formulator, the problem of poor solublility, especially for BCS-II class compounds, can be successfully solved by enhancing aqueous solubility and dissolution rate.
Various approaches can be attempted to enhance the solubility of poorly soluble Prasugrel Hydrochloride. Different techniques are used to improve the solubility thereby increasing bioavailability of poorly soluble drugs and reducing the erratic absorption7. The approaches include aqueous pH shifted solutions (provided the drug has ionizable groups), micronization, solubilization using co-solvents, solid dispersions, oily solutions, and salt formation. These techniques for solubility enhancement have certain limitations. Excepting for pH shifted solutions, large amounts of excipients are needed to improve the solubility of the drugs using other techniques. A classical solubility approach commonly used is micronization8. In micronization, a coarse drug powder is milled to an ultrafine powder with a mean size being typically in the range of 1-10 µm. The solubility of the drugs is enhanced by increase in the surface area. However, new drugs such as Prasugrel Hydrochloride are so poorly soluble that micronization technique cannot solve the solubility issue. The various other techniques used to enhance the solubility and thereby bioavailability includes: microemulsions, nanoemulsions, liposomes, cyclodextrins. Excepting for addition of cyclodextrins, the other techniques are not practically possible because these formulations are poorly stable and also their manufacture is difficult9. Because of these reasons, there is an urgent need to find an efficient way of enhancing solubility and dissolution rate of poorly soluble drugs such as Prasugrel Hydrochloride. Recent addition to this list includes nanotechnology based nanosuspensions10. Nanotechnology can be used to solve the problems associated with various other approaches described. It is also a practically feasible methodology. Currently nine nanosuspension based formulations are available in US market and all of these formulations are approved by USFDA11. One aim of this study was to prepare Prasugrel Hydrochloride nanosuspensions, cyclodextrin inclusion complexes, SLS drug powders, characterize and evaluate the same for enhancement of dissolution rate and in-vivo absorption in rats. The second aim was to develop IVIVC for the Prasugrel Hydrochloride nanosuspensions that could aid in the further formulation development of nanosuspensions12.
A series of Prasugrel Hydrochloride nanosuspensions were developed altering the two factors: surfactant concentrations and solvent/antisolvent ratio. The oral absorption of Prasugrel Hydrochloride from these formulations was evaluated after a single dose to the rats. Further optimization development of a nano formulation involving alterations in formulation development, preparation process, equipment, batch sizes would require additional animal or human studies. These refinements would delay product development of a nanosuspension. It would be desirable to develop a relationship, referred to as an IVIVC, to predict in-vivo absorption of any refined formulation from in- vitro dissolution test as a surrogate for animal/human testing. Establishment of an in-vitro dissolution test as a surrogate for animal testing/human studies was explored in this study to support PHCl nanosuspension formulation development. Such a development for nanosuspensions was proposed and investigated for the first time 13.
Prasugrel hydrochloride were obtained as a gift sample from Suven Nishtaa. Ethanol, Tween 80 and Sodium lauryl sulphate were obtained from S.D Fine Chemicals Limited. All the other ingredients used were of analytical grade[14].
Preparation of Prasugrel Hydrochloride Nanosuspensions:
Prasugrel Hydrochloride nanosuspensions were prepared by evaporative precipitation method. Ethanol was used as a solvent and tween-80 was used as a surfactant to stabilize the nanosuspension formulation. The drug was dissolved in the solvent (ethanol). The syringe was filled with the prepared solution and injected drop wise into the anti-solvent (water containing tween-80) with constant stirring on a magnetic stirrer. Vacuum was applied while stirring till the solvent evaporates to form the product. Finally the product was filtered and centrifuged at 7000 rpm for 10 min. So as to optimize the nanosuspension formulations prepared, several nanosuspensions were prepared based on different compositions given in Table-1. The two factors: surfactant concentration and solvent-antisolvent ratio were investigated at different levels to study their effect on particle size. Several previous studies indicated that these two factors signficantly affect the particle size of the fabricated nanosuspensions [15,16,17].
Formulation | F1 | F2 | F3 | F4 | F5 |
Prasugrel Hydrochloride (mg) | 100 | 100 | 100 | 100 | 100 |
Distilled water (mL) | 100 | 100 | 100 | 100 | 100 |
Ethanol (mL) | 15 | 10 | 20 | 20 | 20 |
Tween 80 (mL) | 0.2 | 0.2 | 0.1 | 0.3 | 0.2 |
Solvent/antisolvent ratio | 1:6.67 | 1:10 | 1:5 | 1:5 | 1:5 |
Table 1: Composition of Prasugrel Hydrochloride Nanosuspensions
| Formulation | % Drug Encapsulation Efficiency |
| F1 | 83.25 |
| F2 | 92.98 |
| F3 | 79.78 |
| F4 | 89.34 |
| F5 | 88.76 |
Table 2: Drug entrapment efficiency of Prasugrel Hydrochloride Nanosuspensions
| Formulation | Particl Size in (µm) |
| F1 | 0.79 |
| F2 | 0.70 |
| F3 | 1.52 |
| F4 | 0.94 |
| F5 | 1.05 |
Table 3: Particle size determination of Prasugrel Hydrochloride Nanosuspensions by optical microscopy method
Characterization of Various Formulations of Prasugrel Nonosuspensions
Drug-excipient compatibility studies:
FTIR: Fourier Transform Infrared (FTIR) spectroscopy:
FT-IR is used for the drug-excipient compatibility samples of about 5mg was mixed thoroughly with100mg KBr IR powder and compacted under vacuum at a pressure of about 12psi, for 3min. The resultant disc was mounted in a suitable holder in Perkin Elmer IR spectrophotometer and the IR spectrum was recorded from 4000cm-1 to 400cm-1 in a scan time of 12min. these studies were done for Prasugrel Hydrochloride, physical mixture and prasugrel nanosuspension [18].
Characterization of Prasugrel Nanosuspensions:
Percentage yield:
To determine the yield, the weight of microspheres obtained at the end of preparation was determined. The total weight of raw materials used to obtain these microspheres was determined to obtain the theoretical yield.
Percentage yield was then determined using the formula:
Percentage yield = (Practical yield/theoretical yield) x 100
Drug entrapment efficiency:
Drug entrapment efficiency was determined by the dialysis method. Drug entrapment efficiency was calculated for the samples of various prasugrel nanosuspensions. Take a beaker containing 100ml of phosphate buffer medium and keep them on the magnetic stirrer for stirring. 10mg of samples was weighed and taken in a broken test tube containing 10ml of phosphate buffer medium. Bottom end of the test tube was packed with the diffusion membrane. Place the test tube inside the beaker and adjust it such that the test tube is submerged in the beaker containing phosphate buffer medium. Now collect the samples 10 ml from the beaker for every 1hr till 10 hrs and replace it with the fresh medium of buffer.
Entrapment efficiency % = Weight of the drug in nanoparticles x 100%
Weight of the nanoparticles
Particle size:
The mean particle size of nanosuspensions was determined using optical microscope. In this method size of 200 particles was determined by using stage micrometer. The average article size was determined.
Surface morphology
In order to examine the particle surface morphology and shape, scanning electron microscopy (SEM) was used. A concentrated aqueous suspension i.e Prasugrel Hydrochloride nanosuspension was spread over a slab and dried under vacuum. The sample was shadowed in a cathodic evaporator with gold layer 20 nm thick. Photographs were taken using a JSM-5200 Scanning Electron Microscope (Tokyo, Japan) operated at 20 kV [19].
Zeta Potential:
The electrophoretic mobility and zeta potential were measured using a zeta potentiometer (Malvern Zetasizer). To determine the zeta-potential various nanosuspension samples were diluted with KCl (0.1 mm) and placed in the electrophoretic cell where an electric field of 15.2 V/cm was applied. Each sample was analyzed in triplicate.
Solid State stability studies:
Differential scanning calorimetry (DSC):
Differential scanning calorimetry (DSC) is a thermo analytical method used to study thermal transitions involving energy or heat capacity changes. Thermal properties of the powder samples were investigated with differential scanning colorimetry. Approximately 10mg of samples was analyzed in an open aluminum pan and heated at scanning rate of 10oc/min between 0oC and 400oC. Magnesia was used as the standard reference materials. The thermograms of Prasugrel Hydrochloride and for the optimized formula of nanosuspension were determined for solid state stability studies [20].
Powder X-Ray Diffraction (XPRD) analysis:
The drug crystalline state in the polymer sample was evaluated by Powder X-Ray Diffraction (XPRD) analysis. Xray spectra were recorded with X’Pert-PRO multipurpose X-Ray diffractometer (PANalytical, Tokyo, Japan) using Ni-filtered, Cu Ka radiation, a voltage of 45 kV, and a current of 40 mA with a scintillation counter. The instrument was operated in the continuous scanning speed of 4°/min over a 2θ range of 5° to 40°. The samples were grinded using a wedgwood mortar and pestle, placed into the cavity of an aluminum sample holder and packed smoothly using a glass slide. The smallest size nanosuspension was used in the study.
In-vitro drug release studies:
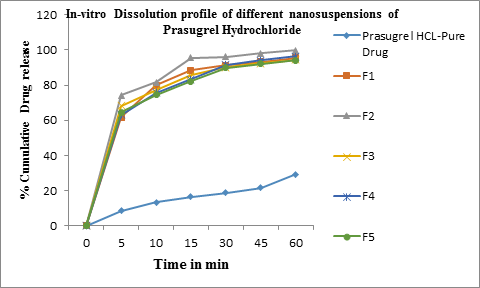
In-vitro drug release experiments were conducted for prasugrel Hydrochloride drug. In-vitro dissolution studies of samples were carried out using USP apparatus-II paddle method by dispersed powder technique. Accurately weighed nanosuspension samples containing 50 mg of drug were added to 500 mL of buffer media at 37 + 0.5°C and stirred at 75 rpm. An aliquot of 10mL was withdrawn at different time intervals. An equal volume of fresh dissolution medium was immediately replaced. The samples withdrawn were filtered by using 0.2µ sterile filter. The amount of drug in the release medium was determined by UV-Visible spectrophotometer. The samples were assayed spectrophotometrically at 254 nm. Citro-phosphate buffer pH 4.0 was used as dissolution medium. This method is based on USFDA recommendations for Prasugrel hydrochloride tablets. The dissolution of nanosuspensions was compared with that of dissolution of equivalent amount of the pure drug. The cumulative amount of drug release over the time period was plotted [21,22].
In vivo Pharmacokinetic Studies in Rats
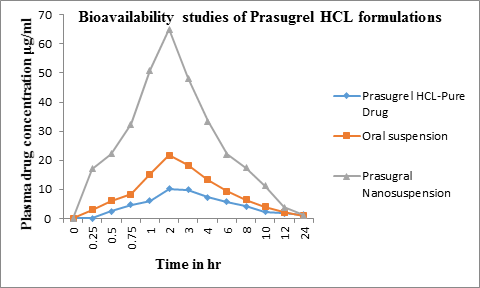
All the animal studies were conducted as per the guidelines of CPCSEA, India. The protocol was approved by Institutional Animal Ethics Committee of Geetanjali College of Pharmacy, Hyderabad (IAEC No. 1648/PO/a/12/CPCSEA). Wistar rats (weighted 180-220 g) were used as experimental animals. Eighteen rats randomly divided into three groups with six rats in each group. Prior to the experimentation the rats were fasted for 12 h with free access to water. The next day, Prasugrel Hydrochloride drug-complex, Prasugrel Hydrochloride nanosuspensions (F2), Prasugrel Hydrochloride oral suspension prepared using sodium CMC as suspending agent were given to the four groups of rats via the oral route. All the formulations contained 50 mg of drug. About 0.5 mL blood samples were collected via the orbit vein at 0.125, 0.25, 0.5, 1, 2, 3, 4, 6, 8, 10, 12 and 24 hr after administration. The collected blood samples were placed in heparinized tube and then separated immediately by centrifugation at 3000 rpm for 10 min and stored at -20ºC prior to the analysis. The plasma samples were then extracted and the prasugrel was analyzed using a LCMS method as described earlier. The method was validated oer a concentration range of 25-10000 ng/mL. The peak areas of all the metabolites was summed and assumed to be the active drug in the plasma that was absorbed. The plasma concentration time profile for all the batches was determined and plotted using excel [23,24].
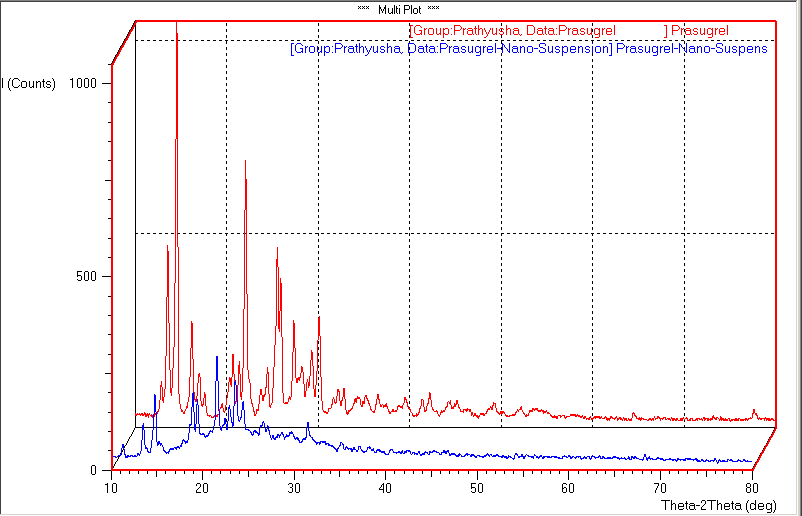
Preformulation studies for prasugrel hydrochloride have been performed to know the drugs physicochemical properties so as to design it to a suitable formulation. Prasugrel hydrochloride is a class-II drug classified under BCS classification with low solubility and high permeability. The physicochemical properties were described. Poor solubility leads to poor dissolution, therefore to enhance the dissolution of the drug, different techniques have been employed such as particle size reduction by forming nanosuspensions which is a novel technique. Nanosuspensions of Prasugrel Hydrochloride (F1-F5) were successfully prepared using evaporative precipitation technique. Several batches of nanosuspsensions altering various parameters were prepared at the initial step. The yield and the particle size were used to draw final conclusions for further studies. Subsequently, it was identified that the surfactant levels as well as solvent:antisolvent ratio were found to have a significant influence on the size. Different parameters were thus altered so as to obtain the smallest particle size of the nanosuspensions. The different batches that were prepared for studying the influence of selected parameters is shown in Table-1. Increase in the concentration of surfactant reduced the particle size. The particle size was also reduced as the solvent/antisolvent ratio was decreased. Based on the process development studies, formulation which had the lowest particle size was considered to be the optimized batch. F2 was considered to be the optimized batch. Further studies were conducted using this batch. The particle size is identified by optical microscopy method and size ranges in micron size from 0.70 to 1.05 µm shown in table-2. SEM was used to determine the particle size and surface morphology of optimized nanosuspension of prasugrel htdrochloride F2 from the figure-1 and 2, it was concluded that the average particle size for all formulations was found to be in nano range of 257 nm to 368 nm. Surface morphology and shape were visualized. The particles were appeared as spheres. From this study it has been concluded that size reduction of particle to nanosize results in enhancing dissolution rate of Prasugrel hydrochloride. The zeta potential of optimized nanosuspension was found to be -30.40 ± 0.12 mV, which indicates the nanosuspension is physically stable which can avoid ostwald ripening. The results of percentage drug entrapment efficiency are shown in the table. 3. From the results shown in the table, it can beinferred that there is a proper distribution of prasugrel hydrochloride in the nanosuspensions. The percentage entrapment efficiency was found to be 79.78% to 92.98%. A maximum of 92.98% drug entrapment efficiency was obtained in the F2 formulation with 1:10 solvent:antisolvent ratio. From, XRPD graphs we can conclude that the crystallanity of the drug was changed in the nanosuspensions in figure 3. The peaks obtained for pure drug was very clear and sharp, the intensity of the peaks was very high when compared to peaks of Prasugrel Hydrochloride nanosuspensions. The peak intensity reduction indicates the reduction in the crystallization nature of the drug. In-vitro drug release from the Prasugrel Hydrochloride and Prasugrel Hydrochloride nanosuspensions in citro- phosphate buffer pH 4 was performed by the dissolution experiment using USP dissolution apparatus II. In dissolution studies, it was observed that the dissolution was higher and 4 fold times for nanosuspensions compared to pure drug shown in table 4 and figure 4. At the end of 1 hr, 99.88% of drug was released with nanosuspensions, while this release was only 29.34% with pure drug prasugrel hydrochloride. The in-vivo study in wistar rats was performed to demonstrate in-vivo bioavailability. DSC studies were conducted with drug cyclodextrin complexes. From the overlay of DSC thermograms, it has been observed that prasugrel is in crystalline nature in figure 5. It exhibited a broad exotherm at 134.4°C and peak onset temperature was found at 117.82°C. For cyclodextrin the DSC thermogram exhibits the peak at 150.29°C and peak onset temperature at 107.83°C. This peak was the characteristic peak for cyclodextrins. The thermograms of the complexes prepared using all the three techniques demonstrated characteristic peaks of both PHCl and cyclodextrins. However, the intensity of drug peaks was lower compared to pure drug. In co-evaporation method an additional sharp endotherm peak was found at 197.78°C. This could be because of conversion of PHCl to its base form during the process of complexation. From all the DSC peaks we can conclude that the inclusion complex was formed between the PHCl and HPβCD. It has indicated that the amorphous form of drug prasugrel hydrochloride has been changed to its crystalline form. Second evidence is obtained from FTIR results (spectrums not shown). Pure HPβCD has characteristic IR peaks at 3421 cmˉ1 (H-S moiety), 2436 cmˉ1 (pyridine group), 1757.21cmˉ1(C=O), 1689.70cm-1(C=O), 1492.95cm-1(C-C aromatic stretch), 1232.55cm-1(C-O), 758 cm-1(C-H aromatic ring). Pure HPβCD has characteristic peaks at 3421.83cm-1, 2931.90 cm-1, 1508.38cm-1, 1082.210 cm-1. In physical mixture, kneading and co- evaporation IR graphs both drug and cyclodextrin peaks were seen, however peak intensities were reduced indicating an interaction between two compounds. The results indicate a possible complex formation because of the disappearance of pyrrolidine group in IR spectra. Stretching bands at 1000-1200 cm-1 indicates the presence of ether group(C-O). The intensity of this peak appeared stronger for co-evaporation and kneading methods. Stretching region of hydroxyl group (O-H) appeared at the range of 3600-3200cm-1. Carbonyl group (C=O) appeared at the range of 1650-1620 cm-1. Inclusion complex bands formed by co-evaporation and kneading methods were shifted to higher wave number indicating the formation of complexes. FTIR results suggest the formation of inclusion complexes of PHCl with cyclodextrins with all the three techniques. It indicates that the drug prasugrel hydrochloride is compatible in the formulation. After orally administration of pure drug, oral suspensions and nanosuspensions, the plasma concentrations were obtained and compared in table 5 and figure 6. The plasma concentration profile for nanosuspension represented significant improvement in drug absorption compared with the oral suspension and pure drug prasugrel hydrochloride. The Cmax and area under the curve (AUC) 0-24 h values of nanosuspensions were approximately 4-fold, greater than that of the oral suspension, indicating a remarkable improvement in the oral absorption of prasugrel nanosuspensions. The Cmax was found to be 69.54 μg/mL at 2hr for nanosuspension. The enhancement in oral bioavailability can be attributed to the adhesiveness of the drug nanosuspension, increased surface area due to reduction in particle size, increased saturation solubility, leading to an increased concentration gradient between the gastrointestinal tract lumen and blood, and increased dissolution velocity. The different pharmacokinetic parameters are calculated given in table 6.


Time in min | Pure drug(PHCL) | F1 | F2 | F3 | F4 | F5 |
0 | 0 | 0 | 0 | 0 | 0 | 0 |
5 | 8.45 | 61.63 | 74.25 | 68.32 | 63.25 | 64.64 |
10 | 13.25 | 79.94 | 81.54 | 77.23 | 75.67 | 74.65 |
15 | 16.54 | 88.32 | 95.43 | 85.63 | 83.22 | 82.34 |
30 | 18.67 | 91.23 | 96.18 | 90.12 | 91.53 | 89.79 |
45 | 21.64 | 93.22 | 98.13 | 92.35 | 94.25 | 92.13 |
60 | 29.34 | 95.67 | 99.88 | 94.56 | 96.35 | 94.12 |
Table 4: In-vitro Dissolution profile of Prasugrel Hydrochloride nanosuspensions

Time in hrs | Pure drug μg/mL) | Oral Suspension (μg/mL) | Nanosuspension (μg/mL) |
0 | 0 | 0 | 0 |
0.25 | 0.10 | 3.01 | 16.99 |
0.5 | 2.45 | 6.01 | 22.26 |
0.75 | 4.66 | 8.32 | 32.32 |
1 | 6.12 | 15.11 | 51.01 |
2 | 10.10 | 21.69 | 65.10 |
3 | 9.82 | 18.32 | 48.12 |
4 | 7.32 | 13.21 | 33.33 |
6 | 5.63 | 9.37 | 22.12 |
8 | 4.12 | 6.33 | 17.45 |
10 | 2.31 | 4.03 | 11.23 |
12 | 1.81 | 2.13 | 3.89 |
24 | 1.20 | 0.85 | 0.99 |
Table 5: Plasma drug concentrations of pure drug, oral suspension and Prasugrel nanosuspension:
S.No | Parameter | Value | ||
Pure drug | Suspension | Nanosuspension | ||
1 | AUC | 80.51 μg.hr/mL | 133.07 μg.hr/mL | 347.75 μg.hr/Ml |
2 | K | -0.126/hr | -0.098/hr | -0.095/hr |
3 | t1/2 | 5.51 hrs | 7.06 hrs | 7.24 hrs |
4 | Cmax | 10.12 μg/mL | 23.14 μg/mL | 69.54 μg/Ml |
5 | tmax | 2.1 hrs | 1.98 hrs | 1.59 hrs |
Table 6: Pharmacokinetic parameters of various Prasugrel Hydrochloride formulations
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
