
Short Communication | DOI: https://doi.org/10.31579/2640-1053/072
The Totteridge Institute for Advanced Studies, The Grange, Grange Avenue, London N20 8AB UK.
*Corresponding Author: Patrick A Riley, The Totteridge Institute for Advanced Studies, The Grange, Grange Avenue, London N20 8AB UK.
Citation: Patrick A Riley. (2021) Overcoming the problem of the lack of an immune response to cancer: A possible haptogenic approach to cancer immunotherapy. Cancer Research and Cellular Therapeutics. 5(1); Doi: 10.31579/2640-1053/072
Copyright: © 2021 Patrick A Riley, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium,provided the original author and source are credited.
Received: 25 January 2021 | Accepted: 25 February 2021 | Published: 01 March 2021
Keywords: epigenetics, malignancy, melanoma, haptogesis, immunology
Cancer cells possess a number of unusual features, most of which are explicable in the light of the theory of epigenetic carcinogenesis. This includes the remarkable failure of malignant cells to evoke an immunological response from the host which is ascribed to their deviant behaviour resulting from anomalous expression of normal gene products. Given this background a possible approach to eliciting a specific anti-cancer immune response is proposed which involves selective haptenation of an identifiable target protein.
The theory that the fundamental pathogenesis of cancer involves initiation by somatic mutation of genes that are responsible for the copying of the epigenetic pattern of differentiated cells during mitosis [1-3] is consistent with the evidence of major epigenetic abnormalities associated with cancer [4-6] and is remarkably powerful in enabling several unique and puzzling features of malignancy to be explained.
Cancer incidence reflects size and proliferation rate of tissue stem cells
Because the proposed defect is envisaged as resulting in inaccurate copying of the epigenetic pattern during mitosis the association with the proliferation rate of stem cells is explained. Malignancy is rare in slowly proliferating tissues but relatively common in rapidly renewing tissues such as epithelia and the cancer incidence in proliferating tissue is related to factors that control the rate of turnover, such as the effect of hormones on tissues like the breast [7]. Furthermore, there is evidence to show that the cancer incidence is a function of the turn over and number of stem cells that comprise a tissue [8]. All these observations are consistent with the existence of an abnormality of the type described by the theory of epigenetic carcinogenesis.
Initiation is consistent with the involvement of a limited number of somatic mutations
The epigenetic carcinogenesis mechanism is consistent with a limited number of initiating somatic mutations as embodied by the model of cancer incidence of Armitage and Doll [9]. It seems likely that the initiating defect arises in some essential epigenetic replication mechanism such as the copying of the DNA methylation pattern [10-13] and may possibly also involve failure of some quality control process such as the p53 mechanism [14-17]. Thus, assuming homozygosity, as little as 4 mutations could suffice to generate an initiated cell as suggested by mathematical modelling [18, 19].
The progressive nature of the basic error permits the explanation of multiple anomalies
Epigenetic carcinogenesis explains the observation of the multitude of genes that have been shown to be expressed in cancer cells. Malignancy is associated with a remarkably wide range of alterations in genetic expression including over- and under-expression of products that are normally associated with the tissue of origin such as the many oncogenes and tumour- suppressor genes that have been observed in various cancers [20], but also the production of proteins that are normally silenced. These observations are consistent with the proposed defective epigenetic control of gene activation, including the loss of the specific gene silencing pattern found in differentiated cells.
Epigenetic carcinogenesis accounts for the genomic instability which is a fundamental feature of cancer cells
The cytological diagnosis of cancer rests on a number of unusual criteria. The features of malignancy include cellular enlargement, increased nuclear/cytoplasmic ratio, nuclear hyperchromasia, prominent and large nucleoli, abnormal distribution of nuclear chromatin, the presence of abnormal mitoses, as well as nuclear membrane abnormalities and cellular and nuclear pleomorphism. These criteria essentially reflect various aspects of chromosomal instability (CIN) which include chromosome doublings, chromosomal fragmentation and other abnormalities occurring during mitosis which result in large-scale chaotic changes to the genome of cancer cells [21]. All these characteristics derive from abnormalities in the structure of the chromatin. Chromatin is the ultimate carrier of heritable epigenetic information. The packaging of chromatin is dependent on the structure of nucleosomes mediated through modification of histones. This in turn is directed by the local pattern of DNA methylation, which is thus the primary determinant of chromatin architecture [22, 23]. Therefore, the proposed defect in epigenetic copying postulated by the theory of epigenetic carcinogenesis provides a mechanism to account for this feature.
The theory explains the apparent hypermutability of malignant cells
Given that the primary defect envisaged is one that results in error-prone epigenetic copying it would be anticipated that new faults can arise at each mitosis. Such a process inevitably generates an expanding spectrum of abnormalities as the affected clone proliferates and simulates the behaviour characterised as hypermutation [24,25] and suggests an evolutionary progression in which the most successful variants, such as those cells with more rapid growth or exhibiting invasive and metastatic properties, are selected which is consistent with the observed pattern of malignancy.
The notable absence of an immunological response to cancer
As noted by Burnet [26], given the extensive range of abnormalities associated with cancer cells, it is remarkable that there seems to be no general immunological response by the host to their presence and this is a matter that has provoked a great deal of comment. A great deal has been written about the possibility that part of the malignant syndrome includes the ability to interfere with the immune response and much effort has gone into investigating the various immune ‘checkpoints’ that enable malignant cells to escape detection by antibodies or cytotoxic T-cells [27]. But it follows logically from the epigenetic carcinogenesis theory which proposes that the emergence of abnormal properties is due to the anomalous pattern of expression of normal genes, a phenomenon which would not be expected to elicit an immune response. Some success has been demonstrated by the action of synthetic antibodies against a mutant form of the Braf oncogene [28], but this does not address the fundamental problem.
From an immunological point of view, the difficulty posed by the notion that the abnormalities exhibited by cancer cells derive from the anomalous pattern of expression of genes and not due to the abnormality of the genes themselves, is that it lacks a process that renders the products of the abnormally expressed genes immunologically recognizable. To bring this about requires a means of generating neo-antigens that will render the malignant cells ‘visible’ to the immune system.
Selective Haptenation as a general approach to cancer immunotherapy
One possible solution to this quandary is to selectively haptenize one or more gene products that are constitutively expressed in malignant cells. Targets could be products implicated in the faulty epigenetic processes involved in rendering the cells abnormal, or one of the re- expressed gene products that endow the cells with their malignant potential. At present these targets remain obscure but the possibility of engaging the attention of the immune system by selective haptenation has been demonstrated in melanocytes as detailed below.
The initial studies emerged from work in identifying the mode of action of externally applied agents that resulted in skin depigmentation. It had been shown by Oliver et al. [29] that contact with the monobenzyl ether of hydroquinone resulted in marked local depigmentation. A subsequent series of studies demonstrated that a series of phenols possessed a similar action, that the effect was dependent of their oxidation by the melanocytic enzyme tyrosinase, and that the depigmentation was the result of the elimination of melanocytes from the affected skin [30-35]. Initially it was considered that this was due to a direct melanocytotoxic action of the quinone oxidation product of the active agents.
The tyrosinase-specificity suggested a possible therapeutic strategy for the treatment of melanoma and a preliminary clinical trial was instigated in which 4-hydroxyanisole (4HA) was infused intra-arterially. Morgan et al. [36] reported a clinical pilot study in which 4HA was administered to a group of melanoma patients with widespread disease unresponsive to other therapies. Despite the high doses administered, the acute responses were disappointing, suggesting that there was negligible direct cytotoxic action [35]. However, longer term follow-up showed that 45% of cases showed some degree of regression of tumours in the infused zone. There was no evidence of a generalised tumour response or of any distant cutaneous or ocular depigmentation. In one case there was complete regression of secondary tumours in the infused limb. This patient, with multiple recurrences at the site of a skin graft on the left leg, received two courses of 4HA (100 and 84 G by intra-femoral infusion) with a 4-week interval and, when seen 4 weeks after the second infusion, was tumour-free [37]. This delayed reaction would be explained by the initiation of an immune response specific to tyrosinase-expressing cells exposed to 4HA.
It had been observed that the loss of epidermal melanocytes resulting from the local application of depigmenting phenols that the absence of melanocytes was associated with an increase in epidermal Langerhans cells [38] and it was proposed that this indicated the involvement of an immune response. Further investigations led to the proposal of an immune mechanism to account for the loss of melanocytes based on the haptenation of melanogenic proteins.
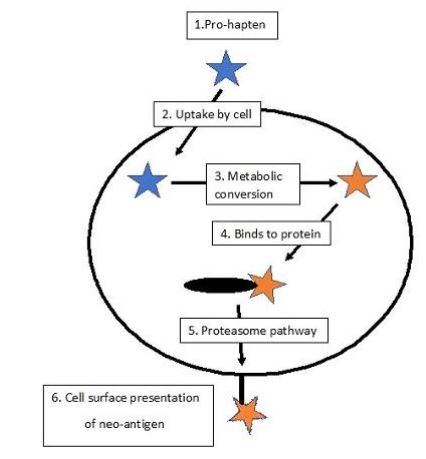
In brief, the proposed mechanism involves the oxidation by tyrosinase of the depigmenting phenol forming the ortho-quinone derivative. These compounds are known to be highly reactive and undergo facile nucleophilic addition reactions [39] binding to adjacent proteins including the enzyme itself. It has been demonstrated, using tritiated 4HA, that the product is covalently bound to tyrosinase [40]. It is proposed that the haptenized proteins are processed by the proteosomal degradation pathway and the neoantigenic products exposed at the cell surface [41]. Here they are recognised by local antigen-presenting cells such as Langerhans cells and this initiates an immune response that includes cytotoxic T-cells which eliminate the melanocytes bearing the haptenic antigen. It has been argued that this is the pathological process underlying vitiligo [42] which is regarded as a manifestation of an autoimmune disease initiated by the haptenation of melanosomal proteins. Since the majority of melanomas express tyrosinase activity, such a targeted haptenation mechanism would account for the results obtained in the preliminary clinical trial of the anti-melanoma treatment and constitutes the mechanistic basis of the melanocyte-directed enzyme prodrug therapy (MDEPT) [43, 44]. The general process is outlined in Figure 1.
This approach suggests that a similar haptenation process could be employed as a therapeutic modality where a suitable expressed enzyme or specific protein target can be identified in the malignant cell population. Spontaneous regression of widespread malignancies, although a comparatively rare occurrence, might suggest that such specific haptenation does take place, since a generalised immune response cannot be explained by a mutation that would affect all the clonal variants present in metastatic cancer. Thus, it is suggested that targeted haptenation could furnish a general means of eliciting anti-cancer immunity and might prove practical as a possible therapeutic strategy.
I am grateful to my colleagues in the Quintox Group and to John Vince, Roger Dean and Mark Burkitt for encouragement, help and advice. I thank Charles Harding for much useful discussion and helpful comments concerning the mathematical modelling. The Totteridge Institute for Advanced Studies is in receipt of a grant from the Melanoma Research Foundation.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
