
Case Report | DOI: https://doi.org/10.31579/2690-4861/035
Neonatology department, Faculty of Medicine, Assiut University, Egypt
*Corresponding Author: Ahmed Mohammad Bakhit, Neonatology department, Faculty of Medicine, Assiut University, Egypt.
Citation: A M Bakhit, M K Moustafa , M S Elyan, E W U Magfory. (2020) Otocephaly:Agnathia Microstomia Synotia Syndrome-A Case Report. International Journal of Clinical Case Reports and Reviews. 3(2); DOI: 10.31579/2690-4861/035
Copyright: © 2020 Ahmed Mohammad Bakhit, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 21 July 2020 | Accepted: 27 August 2020 | Published: 03 September 2020
Keywords: agnathia; antenatal diagnosis; otocephaly
Otocephaly is characterized by aplasia or hypoplasia of mandible, a small oral fissure and low lying ears at the level of neck usually meeting in the midline. This is due to the failure of migration of the ventral part of the first brachial arch. This rare lethal non familial condition occurs in 1 in 70,000 births. This is the first reported case of otocephaly in Tema city, Sohag governorate, Egypt as per our best knowledge, a baby girl born to non-consanguineous parents, who was unable to survive a long time after birth. This case is being reported because of its rarity.
A 40yrs old female patient p1+o, 1 living male , 2ndry infertility 10 years, Previous failed ICSI, pregnant +_ 34 weeks , by ICSI (2nd trial). She admitted to a private centre for Women, Tema, Sohag, Egypt due to severe abdominal pain. There is no consanguinity, there was no exposure to teratogens or infection during the pregnancy. Dating scan was done and cervical cerclage was put at 14th week of gestation. Polyhydramnios was noted on ultrasound scan at 24th week of gestation.
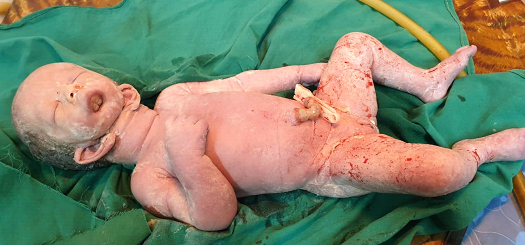
Detail ultrasound scan on this admission revealed a single live fetus with an amniotic fluid index of 43 cm and an estimated fetal weight of 2250 g. There was no definite fetal stomach noted which led to a suspicion of a possibility of esophageal atresia. Baby girl was born by cesarian section with birth weight of 2500 g. Examination of the baby revealed downwards slanted eyes, hypertelorism, very small oral aperture (Microstomia) with blind ended pouch confirmed by inability to pass the orogastric tube, absence of mandible (Agnathia) and external ears displaced ventromedially in the neck close to the mid line (Synotia) (Figure 1). Rest of the clinical examination was normal. Baby died soon after the birth without any active resuscitation, considering the gross abnormalities. Autopsy was advised but refused by the parents. Mother was discharged on following day after counselling with family planning advices.
Discussion
Otocephaly is a rare lethal syndrome of microstomia, agnatia and ear anomalies. Other anomalies associated are holoprosenxcephaly, cephalocele, dysgenesis of corpus colossus, atresis of third ventricle, skeletal, genitourinary, cardiovascular system and endocrine gland hypoplasia [1, 2]. The incidence of otocephaly which is a rare idiopathic malformation in humans is 1 per 70,000 new-borns [3]. Every pharyngeal arch has a core of cartilage, unsegmented mesoderm, an artery from aortic arch and a nerve that enters it from the brain stem carrying motor fibers to supply the skeletal muscle derived from the mesoderm [1]. Arrest in the development of the first branchial arch due to an insult to the neural crest cells has been proposed to be the cause of this condition, which may be induced by chromosomal mutation or teratogens. Otocephaly, with associated anomalies are considered lethal due to severe respiratory dysfunction. Otocephaly is usually suspected on radiological antenatal checkup specially during the third trimester when it is impossible to visualize the mandible and ears are in a very low and in medial position [2,4]. Polyhydramnios may be the presenting feature during pregnancy [3]. In Sri Lanka the terminationof the pregnancy is only considered if the life of the mother is in danger due to the pregnancy, so the antenatal identification of such cases over helps the parents to prepare for the delivery. The syndrome complex of otocephaly is divided into four types: 1. Isolated agnathia;
2. Agnathia with holoprosencephaly;
3. Agnathia with situsinversus and visceral anomalies;
4. Agnathia, holoprosencephaly, situsinversus and other visceral anomaliesm [5].
The case we describe belongs to type 2. Otocephaly anomaly shows spectrum of various manifestations ranges in severity from severe micrognathia as a part of the robin sequence to cyclopiaholoprosencephaly complex invariably associated with fetal death [6]. There are few proposed teratogenic effects as the cause for otocephaly such as exposure strepnigrin antibiotics and trypan blue, theophylline [7]. In our case there was no such exposure identified. The genetic basis of otocephaly is still largely unclear. Even though otocephaly is a lethal syndrome there are cases on successful management of otocephalic babies after birth. Enteral feeding was successful in such newborns with isolated agnathia with nasogastric tubes [6].
Prenatal diagnosis of this lethal condition is possible, and it should be considered as one of the differential diagnosis for polyhydramnios. Poor prognosis of this condition should be discussed with parents prior the delivery. There is still no solid evidence to the cause of this rare condition.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
