
Review Article | DOI: https://doi.org/10.31579/2690-4861/728
Laboratory of Population Ecology, A.N. Severtsov Institute of Ecology and Evolution of the Russian Academy of Sciences, Moscow, Russia.
*Corresponding Author: Victor V. Suntsov, Laboratory of Population Ecology, A.N. Severtsov Institute of Ecology and Evolution of the Russian Academy of Sciences, Moscow, Russia.
Citation: Victor V. Suntsov, (2025), Origin and Evolution of the Plague Microbe Yersinia Pestis: Molecular Genetic and Ecological Scenarios, International Journal of Clinical Case Reports and Reviews, 24(3); DOI:10.31579/2690-4861/728
Copyright: © 2025, Victor V. Suntsov. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 04 March 2025 | Accepted: 14 March 2025 | Published: 17 March 2025
Keywords: yersinia pseudotuberculosis o:1b; yersinia pestis; marmota sibirica; speciation; biocenotic changes; ecological scenario; evolutionary model
It is known that the direct ancestor of the ‘blood’ plague pathogen – the microbe Yersinia pestis – is the saprozoonotic psychrophilic causative agent of the Far Eastern scarlet-like fever (FESLF) Y. pseudotuberculosis O:1b, and the divergence of these species occurred in the recent historical past, no earlier than 30 thousand years ago in the central regions of Asia without any human involvement. These facts indicate certain changes in the habitat of a certain population (clone) of the FESLF pathogen in the region of Central Asia or adjacent regions, which caused the speciation of Y. pestis. Reconstruction of the plague microbe speciation and evolution (its phylogeny/phylogeography) is based substantially on the molecular-genetic (MG) typing of strains from the extant natural foci and fragments of ancient DNA extracted from the remains of victims of the infection. However, presented phylogenies are not congruent with facts accumulated by such classic sciences as environmental ecology (ECO), biogeography, paleontology, epizootology, etc. These contradictions require the development of an integrated methodology for studying the history of the plague pathogen, combining both the model of neutral evolution, which is the theoretical basis in the MG approach, and adaptive evolution, which is the basis of the ECO approach. Herewith an ECO phylogeny as less controversial should be used as a null hypothesis in the phylogenetics/phylogeography of Y. pestis and a factual basis for the synthesis of ECO and MG approaches to the problem of the origin and global expansion of the plague causative agent.
Biological and taxonomic diversity of the microbe pathogens is a result of their historical development. Therefore, it is impossible to identify in detail their relationships with hosts and develop effective control techniques and procedures without insight into their evolution. Historical reconstruction of the phylogeny of causative agents is one of the priority tasks in the modern medical and biological science. At present, phylogenetic reconstructions of any taxa are inconceivable without the use of advanced molecular-genetic (MG) methodology. Meanwhile, the MG approach to the phylogenetics of pathogenic microorganisms is still too young to be considered perfect. Despite undoubtedly great achievements of this approach in diagnosing microorganisms and describing their biological diversity, its conclusions in reconstructing phylogenies are sometimes uncertain and unconvincing. In the past decades, MG approach dominated in the development of MG phylogenetics of microorganisms; however, overestimation of its achievements and underestimation of its succession are evident. The past is hidden by a thick veil of time, and all conceivable techniques, approaches and methods would be useful and necessary for the reconstruction of historical events. Comprehensive scientific experience and knowledge accumulated over the centuries are in demand. In the earliest days of phylogenetics, E. Haeckel proposed to use the method of triple parallelism for reconstruction of the animal phylogenies. The method implied consolidation of the morphological, paleontological and embryological data. Reciprocal verification of phylogenetic inferences based on the use of different approaches and methods greatly improves confidence in their reliability. Such comprehensive methodological approach is also required for the reconstruction of phylogeny of the plague causative agent. Dominating MG approach, when taken individually, still looks imperfect and not self-sufficient.
Phylogeny is a history of taxa, the roots of which in the overwhelming majority of cases have long since became extinct or are unknown. The ancestors have to be ‘figured out’ and ‘identified’ based on the characteristic traits of the extant organisms and fossils. The situation with the plague microbe Yersinia pestis is different. The latest MG data indicate that its phylogeny is inherently a reflection of the recent process of speciation and development of the extant intraspecific forms. To date, intraspecific ecological (by the main host), biochemical, genetic, and molecular diversity of the plague microbe and its various intraspecific forms (subspecies, genovariants, biovars, ecotypes, plasmidovars, etc.) have been described in sufficient detail. The fragments of ancient DNA of the plague microbe have been discovered in the remains of human victims of past pandemics. However, despite great achievements in the study of the plague microbe, the patterns of its speciation and subsequent intraspecific evolution remain obscure. The molecular characteristics of intraspecific forms say little about the strict order of their formation in time. The MG hypotheses are largely contradictory and detached from natural and historical events [1]. Meanwhile, since the time when the plague microbe was discovered about 130 years ago, a great wealth of general biological and medical knowledge has been accumulated. This available information is not only useful; in the light of new data obtained by the MG approach, it is indispensable for an appropriate reconstruction of the history of this evolutionarily young pathogen and for the improvement of the MG methodology of phylogenetic constructions.
We address the potential of reconstruction of the plague microbe phylogeny by molecular and genetic methods and show their fundamentally limited capabilities in solving this complex problem. We argue that the prospect of constructing a real phylogeny lies in the integration of ecological (sensu lato) and MG approaches. The ecological approach that we have been developing over the past 20-25 years has not yet been widely discussed in the scientific literature, so we often have to resort to self-citation and refer in the text to our early publications.
Two MG discoveries
The introduction of MG methods into bacteriology in the past two or three decades has led to two cornerstone discoveries in the problem of plague. First, the genetic analysis (study of O-antigen and conservative genes thrA, trpE, glnA, tmk, dmsA, manB) revealed an apparent direct ancestor of the plague microbe – a pseudotuberculous microbe of the 1st serotype Y. pseudotuberculosis O:1b (family Yersiniaceae/Enterobacteriaceae), a causative agent of Far East scarlet-like fever (FESLF) [2]. This intestinal pathogene is widely distributed in populations of a wide range of invertebrates and vertebrates in the cold regions of Siberia, the Far East, and Central Asia [3, 4]. Hence, the conclusion follows, that the speciation of the plague microbe from a clone of the FESLF causative agent occurred in a certain cold region of Central Asia with numerous natural foci of plague. The most likely candidate is Mongolia with its ultracontinental climate or the nearest vicinity of this country [5]. Second, contrary to the key concept of the ‘Sylvatic Plague’ theory about the great antiquity of this infection, the ‘molecular clock’ technique showed its evolutionary youth. According to molecular estimates, the causative agent of this disease emerged no earlier than 30 thousand years ago, i.e., at the turn of the Pleistocene and the Holocene or in the Holocene [6, 7]. Common evolutionary sense implies that this event was caused by environmental changes in the habitats of the FESLF causative agent. It is known that it was the last maximum Sartan cooling 22–15 thousand years ago that resulted in the most extensive climate change and notable disturbances in the North and Central Asian biocenoses. The cooling led to intensive process of speciation and development of new forms of organisms. Natural ranges of many animals and plants in Asia changed in that period. The southern border of the permafrost zone in Asia shifted almost a thousand kilometers to 42°N and reached the Gobi Desert in southern Mongolia [8]. The two aforementioned MG discoveries logically linked numerous facts from various scientific fields (ecology, biogeography, epizootology, paleontology, microbiology, paleoclimatology, etc.) and allowed development of an ecological sensu lato (ECO) scenario of the emergence and Asian expansion of a new highly pathogenic species Y. pestis, which is fundamentally different from MG scenarios [9–12].
MG phylogenies of Y. pestis
Modern phylogenies of the plague causative agent are based on the molecular approach. The most popular methodology operates with the maximum likelihood method and Bayesian analysis. In recent years, single nucleotide polymorphism (SNP) markers are more often analyzed, and the model of uniform neutral evolution is used as the basic evolutionary model (Figure 1a) [12, 13]. An important result of the MG approach is the detection of paraphyletic origin of the cluster of ‘vole’ genovariants/subspecies 0.PE (Pestoides biovar) circulating in the populations of voles (Microtina), the Siberian jerboa (Allactaga sibirica) and Mongolian pika (Ochotona pallasi pricei) in Asia [13–16]. Genovariants/subspecies of the ‘vole’ paraphyletic group 0.PE were shown to originate from an abstract most recent common ancestor (MRCA) at different times and in different geographical regions. Unfortunately, the MG approach provides no explanations for the fact that the 0.PE cluster consisting of so-called ‘non-main’ subspecies 0.PE2, 0.PE3, 0.PE4, 0.PE5, 0.PE7 and 0.PE10 formed as a paraphyletic group, whereas all other known subspecies of clusters or phylogenetic branches 0.ANT, 1.ANT, 1.IN, 1.ORI, 2.ANT, 2.MED, 3.ANT and 4.ANT were combined into a single ‘main’ subspecies Y. pestis pestis, that is, into a single monophyletic group. It is believed to originate from a single root – the population of the causative agent of plague circulating in the populations of Altai marmot (Marmota baibacina centralis) in the Tien Shan. A credible explanation for this paradox, worthy of a more thorough and comprehensive analysis, lies in the ecological approach outlined below.

Figure 1: Phylogenetic trees of Yersinia pestis: (a) – molecular genetic [13]; (b) – ecological/environmental [12] (clarifications in the text). The red arrow (Figure. 1a) shows that the cluster of marmot genovariants 0.ANT belongs to the node of polytomy N07 [17]. The dashed arrow shows the location of the polytomy node N07 on the ecological three-root tree. The dotted arrows show the location of the decomposition of genovariants/subspecies of the ‘vole’ cluster 0.PE on the ecological three-root tree. Dot arrows indicate the locations of genovariants 2.MED1 and 2.MED3 on the ecological tree. *– diversification of genovariants of the 0.ANT cluster. Phylogenetic lines of marmot subspecies/genovariants are outlined in blue.
<!-- /* Font Definitions */ @font-face {font-family:"Cambria Math"; panose-1:2 4 5 3 5 4 6 3 2 4; mso-font-charset:0; mso-generic-font-family:roman; mso-font-pitch:variable; mso-font-signature:-536869121 1107305727 33554432 0 415 0;} @font-face {font-family:Calibri; panose-1:2 15 5 2 2 2 4 3 2 4; mso-font-charset:0; mso-generic-font-family:swiss; mso-font-pitch:variable; mso-font-signature:-469750017 -1073732485 9 0 511 0;} /* Style Definitions */ p.MsoNormal, li.MsoNormal, div.MsoNormal {mso-style-unhide:no; mso-style-qformat:yes; mso-style-parent:""; margin-top:0in; margin-right:0in; margin-bottom:8.0pt; margin-left:0in; line-height:107%; mso-pagination:widow-orphan; font-size:11.0pt; font-family:"Calibri",sans-serif; mso-ascii-font-family:Calibri; mso-ascii-theme-font:minor-latin; mso-fareast-font-family:Calibri; mso-fareast-theme-font:minor-latin; mso-hansi-font-family:Calibri; mso-hansi-theme-font:minor-latin; mso-bidi-font-family:Arial; mso-bidi-theme-font:minor-bidi;} span.rynqvb {mso-style-name:rynqvb; mso-style-unhide:no;} span.hwtze {mso-style-name:hwtze; mso-style-unhide:no;} .MsoChpDefault {mso-style-type:export-only; mso-default-props:yes; mso-ascii-font-family:Calibri; mso-ascii-theme-font:minor-latin; mso-fareast-font-family:Calibri; mso-fareast-theme-font:minor-latin; mso-hansi-font-family:Calibri; mso-hansi-theme-font:minor-latin; mso-bidi-font-family:Arial; mso-bidi-theme-font:minor-bidi; mso-font-kerning:0pt; mso-ligatures:none;} .MsoPapDefault {mso-style-type:export-only; margin-bottom:8.0pt; line-height:107%;} @page WordSection1 {size:8.5in 11.0in; margin:1.0in 1.0in 1.0in 1.0in; mso-header-margin:.5in; mso-footer-margin:.5in; mso-paper-source:0;} div.WordSection1 {page:WordSection1;} -->
One more important conclusion of the MG approach was the detection of a polytomy on the phylogenetic tree (N07, ‘Big Bang’). It was the starting point for phylogenetic branches 1, 2, 3 and 4 on the eve of the supposed second pandemic (‘Black Death’, 1346). That is, it was shown that a host of natural plague foci formed almost simultaneously (star-shaped) in the populations of marmots, ground squirrels and gerbils in the vast areas of Central Asia and Tibet. Moreover, three different subbranches/genovariants 2.ANT3, 3.ANT2 and 4.ANT1 formed in populations of one host species, the Mongolian marmot (Marmota sibirica). The MG approach fails to reveal or even hypothesize the reason for diversity of the plague causative agent at the level of subbranches/genovariants in Mongolian marmot populations. This phenomenon has a credible ecological explanation. Meanwhile, MG phylogenies demonstrate a number of significant inconsistencies and contradictions with ‘non-molecular’ facts, which cast doubt on ‘molecular’ conclusions in general (Figure 1a,b).
First, the MG approach proclaims a two-stage Asian expansion of the causative agent from the supposed center of speciation (Tibet? Caucasus? Altai? Angola? East Africa?) and the initial formation of the primary range of exclusively ‘vole’ plague foci in Asia. This range encompassed vast territories from northeastern China and eastern Tibet in the east to the Caucasus in the west and, possibly, to Angola, where the ‘non-main’ subspecies 0.PE3 Y. pestis angolica was found [18]. ‘Plague-like’ causative agent in the ‘vole’ foci exhibits selective virulence and, in some MG and biochemical properties, is close to its pseudotuberculous ancestor. According to the MG version, on the eve of the first pandemic (‘Plague of Justinian’, 576 AD), a causative agent of the ‘vole’ subspecies 0.PE5 Y. pestis ulegeica that forms natural foci in populations of the Mongolian pika in the Mongolian Altai and the Altai Mountains in Russia, under unknown circumstances penetrated from Mongolia into the Altai marmot population in the Tien Shan. The subspecies developed there in a highly virulent ‘marmot’ biovar Antiqua (genovariant/subspecies 0.ANT1, 0.ANT2, 0.ANT3 and 0.ANT5). Later, following the MG logic, the causative agent of the real ‘marmot’ plague from the Tien Shan spread in a second wave within the former ‘vole’ range in Asia and dispersed across the northern Caspian region, the Middle East and Hindustan [14]. Such a two-stage pattern of expansion of plague within the Asian range does not fit the epizootological concept of the spread of a causative agent from the area of speciation according to the principle of ‘spreading of an oil stain on the paper’ [19].
Second, the crucial drawback of the present-day MG approach is its limited ability to identify the original host of the plague causative agent solely by statistical methods. Identifying the original host of the plague pathogen is the key to understanding its history. Knowledge of the original host makes it possible to reconstruct its history at the population-species level without resorting to complex MG computational statistical technology. To date, the genetic and nucleotide structures of the causative agent from the majority of natural foci known in the world have been studied. Its most ancient genovariants/subspecies have been identified. However, MG methods do not allow us to indicate populations of the particular rodent species in which the original population of the plague microbe was formed. The MG approach reconstructs only the emergence of the abstract common ancestor (MRCA) of Y. pestis from the FESLF clone. Neither its host environment, nor population characteristics are discussed. A featureless MRCA is characterized only statistically by marker traits that are not determined by ecological functions. At the same time, it is indisputable that new species originate in the bowels of ancestral populations, so the MRCA must represent a certain original subspecies or genovariant of the plague microbe. But for now it is still unclear whether MRCA should be classified as a mature species and initial subspecies of Y. pestis, or a transitional nameless form Y. pseudotuberculosis/pestis. The MG approach clearly indicates the ancestral species (unfortunately, not its specific original population/subspecies) on the phylogenetic tree of Y. pestis. However, it fails to reveal any unique features of the population-genetic speciation process and, main, to specify the original host of the plague microbe. In this case, the MG approach cannot be considered self-sufficient for a reliable reconstruction of the history of the plague causative agent, and the model of neutral evolution seems to be not reliable for the reconstruction of its phylogeny.
Third, the MG approach in phylogenetics of the plague microbe has two components: molecular and genetic. These components are not in harmony with each other, they are alternative and antagonistic. Molecular methodology operates with ‘phylogenetic’ nucleotide markers designated as evolutionarily neutral. ‘Neutral’ model of evolution characterizes the gradual transformation of a taxon under the impact of genetic drift. Analysis of the chosen neutral characters determines the topology of the phylogenetic tree of Y. pestis. At the same time, the genetic methodology operates with ‘adaptive’ traits: the plague microbe has acquired specific factors of virulence, transmission and communication; these factors are encoded by particular genes and gene complexes [20]. Moreover, the genetic approach to Y. pestis phylogenetics proclaims acquisition of specific properties during the ‘saltation evolution’. This evolutionary process implies the horizontal transfer of genes, primarily those of the virulence plasmids pFra and pPst, from the environment or from other microorganisms, as well as single-act deletions and inactivations/pseudogenizations of gene structures that have lost their functions in a new environment [21, 22]. The question is why revolutionary adaptive genetic changes in the plague microbe, which determined its history, are not reflected in the topology of the phylogenetic tree. If we follow the evolutionary logic, the analysis of ‘phylogenetic’ and ‘adaptive’ traits used in reconstruction of Y. pestis phylogeny requires coordination in methodology. Reconstruction of the history of the ‘young’ causative agent of plague should not be restricted to the analysis of only neutral nucleotide ‘phylogenetic’ traits. ‘Adaptive’ features are more indicative in this case; they should be definitely used or even play the main role in the construction of the phylogenetic tree topology.
‘Ancient’ plague foci
Fossil taxa potentially play a key role in reconstructing the history of studied phylogenetic groups, as they represent the roots and early branches of phylogenetic trees. ‘Fossil (archaeological) taxa’ and ‘ancient’ plague foci of Y. pestis are reconstructed from DNA fragments extracted from the remains of human victims of the first and second pandemics and materials found in the earlier burial sites [23–25]. Since some DNA fragments discovered in Europe are close in their nucleotide structure to homologous fragments of pseudotuberculous microbe, the ‘archaeological’ taxa/subspecies Gok2, LNBA and other proposed on the basis of these fragments are placed at the root of phylogenetic trees of Y. pestis. No modern genotypes of the plague microbe with molecular markers homologous to ‘archaeological’ ones have been found, and the ‘archaeological’ taxa/subspecies are considered extinct. However, no environmental conditions convenient for the natural foci of plague are known for post-glacial Europe in the Holocene. The region was lacking arid landscapes populated by numerous burrowing rodents, which were massively parasitized by fleas [26]. Numerous original natural foci were confined to the vast arid Sahara-Gobi zone, but that zone did not stretch into Europe. Therefore, no sound ecological and epizootological grounds can support any natural lineages in the phylogeny of the plague microbe, which became extinct after extinction of the hosts or vectors of the pathogene. The extinct foci of plague in Europe were undoubtedly synanthropic. They had anthropogenic (imported) origin and were most likely supported by stable extant subspecies of the causative agent [26].
Imperfection of the MG-approach
The MG approach to the reconstruction of the history of the plague pathogen relies on the theory of neutral evolution and the corresponding phylogenetic methodology as a theoretical basis. However, the causative agent of plague is unique in the genus Yersinia and the family Yersiniaceae, which combine pathogens of intestinal infections: plague is the only vector-borne infection in these taxa. The unique origin and evolution of the plague pathogen are largely characterized by adaptive (not neutral!) traits: it is the adaptive traits that most clearly characterize the history of the plague microbe. Accordingly, the evolutionary model, along with neutral ones, should provide for adaptive processes, and in phylogenetic reconstructions in addition to molecular and genetic characteristics, should also use ecological (adaptive) ones. In addition, in the problem of reconstructing the history of the plague microbe the methods of analysis widely accepted in molecular phylogenetics do not quite correspond to the object of research. It is known that the causative agent of FESLF is a direct ancestor of plague microbe, and that the approximate time of divergence of plague and pseudotuberculosis microbe is no earlier than 30 thousand years ago. That is, speciation and intraspecific divergence of the plague pathogen took place in the recent evolutionary and historical past in an (almost) modern biogeocenotic environment, in an environment that has an (almost) modern appearance, when modern species and subspecies of rodents-hosts and fleas-vectors of the plague microbe and arid landscapes within (almost) modern their boundaries already existed. In this case, the reconstruction of the history of the plague pathogen is to a greater extent a problem not of phylogenetics, but of phylogeography, which, in addition to molecular and genetic concepts, operates with such ecological concepts as subspecies, geographical population, range, territorial expansion, direct relatedness, gene flow, and many others. From the point of view of the ecological approach, summarized below, some of the methods of the MG approach for solving the history of plague seem to be not fully justified. For example, the CO92 strain isolated from a sick person infected from a domestic cat in the United States is chosen as a reference for ‘calculating’ the degree of kinship of all the studied genovariants/subspecies of the plague microbe, or the strain YP32953 Y. pseudotuberculosis O:1b isolated in France from a sick patient is used as an outgroup (nearest-neighbor) to characterize the root phylogenetic tree. These strains had a long anthropogenic history, not without human participation they got to the United States and France. As a reference strain and a representative of the outgroup, it is more reliable to select strains from wild rodent populations living in Central Asia, the most likely area of origin of plague) [5, 11, 12, 19].
Mongolian marmot is an original host of plague
In case the plague microbe has emerged recently in an almost modern biogeocenoses, an ecological, i.e., adaptationist approach is of particular value for phylogenetic/phylogeographic reconstructions. It provides for a more complete narrative of the history of the plague microbe, rich in biological information. Actually, the reconstruction of speciation processes and intraspecific diversification of the plague causative agent is the prerogative of phylogeography, the methodology of which operates with such concepts as speciation, intraspecific diversification, subspecies, geographic population, range, ecological subniche and migration.
Identification of the original host of the plague causative agent is a cornerstone problem in the reconstruction of its history. It allows matching the significance of ‘adaptive’ (ecological, biochemical, genetic) and neutral ‘phylogenetic’ (IS, VNTR, CRISPR, SNP, etc.) traits/markers in the reconstruction of the evolutionary history of the plague microbe and opens up broad prospects for studies of the patterns of molecular evolution in general. If the real ancestor and the original form of the plague microbe are known, monophyletic groups could be accurately assembled in accordance with the adaptive characteristics and on the basis of biogeographical distribution of the main hosts of the causative agent.
Any speciation process is caused by changes in the habitat of the original population. Consequently, the recent transformation of the clone of the FESLF causative agent into the original population of the plague microbe was caused by a change in its habitat, which led to modification of its life cycle. Causative agent of FESLF inhabits both the external organic matter (the soil, excrements and animal remains) and the digestive tract of a wide range of invertebrates and vertebrates. The habitats of the plague causative agent are the organisms of a rodent (host) and a flea (vector). The key evolutionary event in the genesis of the plague microbe as a new species was the development of the following factors ensuring continuous circulation of the plague microbe: the virulence to the rodent host on the part of the microbe, the immunity to the microbe on the part of the rodent host, and transmission by fleas. In the unanimous opinion of experts, this event occurred in populations of a certain rodent species. A cornerstone issue is the identity of the species. According to unsubstantiated MG versions, the pathogene could be originally hosted by the Nile grass rat Arvicanthis niloticus [27], the Siberian jerboa Allactaga sibirica [13], the Common vole Microtus arvalis [28] and marmots, most likely the Altai marmot [29–31]. However, ecological data definitely indicate that the plague microbe was initially hosted by the Mongolian marmot. No other sufficiently deeply substantiated options have been proposed in the world scientific literature. Therefore, this presumption should be accepted as a null hypothesis for studying the history of plague and its causative agent until new, more substantiated variants of the main host appear.
We argue that the original habitat of the plague microbe was the parasitic system of the Mongolian marmot and the flea Oropsylla silantiewi. A shift of the FESLF causative agent to a new ecological niche and speciation of the plague microbe could have occurred no earlier than 30 thousand years ago. These events were caused by changes in the aforementioned parasitic system under the influence of particular abiotic and/or biotic factors, which needs to be discussed.
The internal environment of homoiothermic animals is regulated by homeostasis mechanisms and remains highly stable. Therefore, there is no reason to believe that in the past 30 thousand years physiological, biochemical and other characteristics of the intestinal habitat of the pseudotuberculous microbe in the body of homoiothermic hosts, including the Mongolian marmot, have undergone any radical changes. In addition, it is necessary to understand why the plague pathogen turned out to be a unique representative of the intestinal microbial family Yersiniaceae/ Enterobacteriaceae? This is the only representative of the family among hundreds that is transmitted not by the traditional alimentary route, but by a transmissible way through flea bites. The question arises: what happened in the nature of Central Asia (or nearest regions) in the recent historical past, which led to the evolutionary transition of the intestinal pathogen into the ‘blood’ of rodents? No causes and mechanisms have been revealed that in the not-too-distant past could have led to colonization of the lymphomyeloid complex of homoiothermic hosts by pseudotuberculous microbes and transmission by overcoming the intestinal epithelial barrier. Therefore, it seems reasonable to accept the alternative that the evolutionary transition of the pseudotuberculous microbe from the digestive tract into the lymphomyeloid complex of the Mongolian marmot was initiated by some external rather then host-dependent factors. According to the ECO scenario, these were two abiotic factors that arose at different times: the aridity and severity of the climate in Central Asia [9–12, 32–34]. The arid climate of Central Asia that gave rise to the development of the species-specific behavior of the Mongolian marmot could be considered a remote abiotic cause. The Mongolian marmot inhabiting arid steppe and mountain-steppe landscapes of Central Asia is the only species that exhibits specific protective behavior. It plugs its wintering burrow with stones and fine soil mixed with its own excrements as a binding material. When building such a plug, a marmot transports construction materials from the toilet chamber in its teeth, and a large amount of excrements together with the FESLF causative agent ends up in the animal’s mouth cavity. It results in extensive accumulation of the causative agent of FESLF in the marmot’s mouth cavity before it gets ready for hibernation. Marmots stop feeding prior to and during hibernation, so the FESLF causative agent does not enter their gastrointestinal tract in this period and the infection process does not develop in hibernating animals. In active marmots, pseudotuberculosis infection proceeds, presumably, like in other mammals.
The Sartan cooling is a trigger for Y. pestis speciation
The accumulation of the FESLF causative agent in the mouth cavity of the Mongolian marmot during hibernation was only a remote prerequisite for speciation of the plague microbe. The actual trigger for the speciation process was the last maximum Sartan cooling in Asia at the Pleistocene-Holocene boundary 22–15 thousand years ago. Within the range of the Mongolian marmot in Mongolia, the maximum cooling resulted in freezing of the soil to the depth of two meters and even deeper [8], which altered the behavior of larvae of the marmot flea O. silantiewi. Under normal conditions, flea larvae feed on detritus in the marmot underground nests. However, with the onset of maximum cooling and consequent deep freezing of the soil, flea larvae shifted to feeding on the blood of dormant hosts in winter and spring. Such a behaviour is still observed in larvae in Southern Siberia and Central Asia nowadays. Hematophagy of marmot flea larvae is not a crucial adaptation; this temporary phenomenon is timed to the cold season and caused by the positive thermotaxis, a primitive behavioral reaction. In winter, the ground freezes to the depth of the hibernation nest chambers of the Mongolian marmot (2–3 m). The flea larvae move from the frozen nest lining to the warmer bodies of dormant animals (the body temperature of a marmot in a torpor state is about 5°C). The larvae stochastically get from marmot’s fur into its mouth cavity, where they injure the mucous lining and feed on oozing blood (facultative hematophagy of flea larvae is often found in fleas of various species that parasitize warm-blooded animals living in cold areas of the world [10]). Thus, facultative behavior of marmot fleas enabled the causative agent of FESLF to penetrate via a traumatic route from the mouth cavity of dormant marmots directly into the blood and cause trivial ‘blood poisoning’ on a population-wide scale. A unique traumatic pathway of FESLF infection in populations of the Mongolian marmot resulted in rapid ‘quantum’ translocation of the intestinal pathogene to a new ecological niche, the lymphomyeloid system of the Mongolian marmot, and its transformation into the plague causative agent Y. pestis [33] (Figure 2).
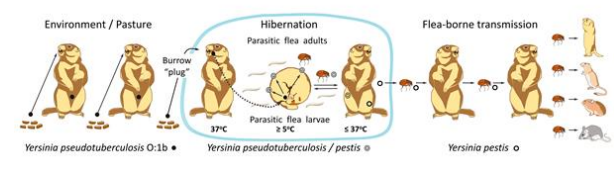
Figure 2: A shift of the Yersinia pseudotuberculosis 0:1b clone to the new ecological niche. The source of FESLF traumatic infection of the Mongolian marmot during hibernation was the excrements, which is accumulated in the toilet chambers in wintering burrows and used to make a burrow ‘plug’ [8, 9]. Body temperature of marmots: in torpor 5оС, in euthermic states 37оС.
Parapatric speciation of the plague causative agent
If the traits of the original host populations in which the intestinal causative agent of FESLF transformed into a blood parasite are known and the evolutionary youth of the plague microbe, as well as its coexistence with a direct ancestor, is considered, the evolutionary history of the plague causative agent can be adequately reconstructed with the use of ecological (sensu lato) methods. The range of the Mongolian marmot in Central Asia is formed by three relatively independent geographical populations. Subspecies M. sibirica sibirica occurs in the Khentei Mountains in eastern Mongolia, Barga and on the western slopes of the Khingan Ridge in northeastern China. M. sibirica caliginosus live on Khangai Highlands in Central Mongolia. M. sibirica ssp., a subspecies not yet described, inhabits the Kharhiraa-Turgen-Mongun-Taiga mountain complex in Tyva and western Mongolia. The Sartan cooling encompassed the entire Central Asia and the plague microbe Y. pestis could be assumed to arise as a species almost simultaneously in three geographical populations of the Mongolian marmot, which gave rise to three geographical populations (subspecies, genovariants) of the pathogene: 2.ANT3 (Khentei, Khingan, Barga), 3.ANT2 (Khangai) and 4.ANT1 (Kharhiraa-Turgen-Mongun-Taiga) [11, 12]. Therefore, the origination of Y. pestis species from three isolated populations of the FESLF causative agent should be considered parapatric (peripatric?) speciation, whereas the ‘explosive’ emergence of genovariants/subspecies 2. ANT3, 3.ANT2 and 4.ANT1 in populations of the Mongolian marmot is reflected in the molecular phylogenies as a node of polytomy N07 (‘Big Bang’) (Figure 1, 3A,B).
Further territorial expansion and host specialization followed independent routes (Figure 3A). As a consequence, Y. pestis represents a paraphyletic group of three holophyletic independent groups, rather than a single monophyletic group of taxa (subspecies). Similarity of three holophyletic groups in the nucleotide markers is associated with parallelisms that arose during the one-way evolution of vicariating populations under (almost) identical conditions. In N07 node, the derivatives of the ‘gerbil-gopher’ genovariants 2.MED1 and 2.MED3 do not form a single monophyletic branch of 2.MED, but are included in different holophyletic groups 2.ANT3 and 4.ANT1 (Figure 1).The homoplastic similarity of holophyletic groups, but not their direct kinship, predetermines the need for a unique ad hoc model of the evolution of the analyzed molecular characters.
Awareness of the nature of the direct ancestor of the plague microbe (intestinal causative agent of FESLF) and the original host (the Mongolian marmot), a well-established divergence from the ancestor in the recent geological past, and the unique position of the ‘blood’ plague microbe among the intestinal inhabitants of the family Yersiniaceae (Enterobacteriaceae) allow us to base the topology of the Y. pestis phylogenetic tree on ecological and biogeographical data. Complex molecular statistical calculations of the historical trajectories of the pathogen, which are redundant in this unique case, can be avoided. Ecological evidence suggests rapid ‘quantum’ speciation of the plague microbe in three geographic populations of the Mongolian marmot. The transition of original subspecies 2.ANT3, 3.ANT2 and 4.ANT1 from populations of the Mongolian marmot to sympatric, semisimpatric and/or adjoining populations of other host species (the Daurian and long-tailed ground squirrels, Mongolian gerbil, Brandt vole, Mongolian pika, Altai marmot) accompanied by transformation into different genovariants fit the principle of ‘spreading of an oil stain on the paper’. Adaptation to new hosts led to differentiation of new microbial subspecies.
The topology of Y. pestis phylogenetic tree proposed by the ecological approach, is fundamentally different from topologies derived from the results of molecular studies. But the history of the plague microbe is invariant. Harmonization of ECO and MG methodologies should be an immediate task of phylogenetic studies of the plague causative agent. The MG approach considers a model of neutral uniform evolution as an evolutionary model. The comparison of SNP traits (or other molecular characters) of all studied intraspecific forms (genovariants, subspecies) of the plague microbe and the ancestral causative agent of FESLF is based on a unified algorithm. However, the speciation and expansion of the plague microbe are essentially different processes. Speciation is a ‘quantum’ population genetic process with the shift of the evolving group (taxon) to a fundamentally different ecological niche and adaptive zone. This process had a macroevolutionary effect. The newly formed species was assigned the taxonomic status of a new species Y. pestis in the family Yersiniaceae (Enterobacteriaceae), but this new species is worthy of the status of a new family of ‘blood parasites’. Expansion is a gradual microevolutionary process with frequent bottleneck effects arising when the pathogene shifts to a new host. Since the macro- and microevolutionary processes in the history of the plague microbe are not identical, the basic model of evolution of characteristic traits in phylogenetic constructions should be an ad hoc model. It should somehow simultaneously reflect various aspects of the evolutionary process: neutral and adaptive, gradual and quantum, macro- and microevolutionary. The prospects for the development of such a combinatorial model are a function of the progress in integration of the achievements of molecular genetic and ecological approaches.
No funding was used.
Ethics approval and consent to participate.
This work does not contain any studies involving human and animal subjects.
The author of this work declares that he has no conflicts of interest.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
