
Research Article | DOI: https://doi.org/10.31579/IJBR-2021/019
*Corresponding Author: Bhaskar Bhadra, Synthetic Biology group, Reliance Corporate Park, Reliance Industries Limited, Navi Mumbai 400701, India
Citation: Abhishek M. Kulkarni , Vikas K. Patel, Akshay C. Chawande, Paul K, Manish R. Shukla, Bhadra B., Dasgupta S. (2021) Optimization and validation of multiplex-PCR method for diagnosis of SARS-CoV-2; International J. of Biomed Research. 1(5); DOI: 10.31579/IJBR-2021/019
Copyright: © 2021, Bhaskar Bhadra, This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 05 May 2021 | Accepted: 12 May 2021 | Published: 17 June 2021
Keywords: SARS-CoV-2, COVID 19, one-step multiplex PCR, structural genes, bioanalyzer, signature sequences
The recent outbreak of COVID 19 pandemic caused by SARS-CoV-2 Coronaviruses claimed millions of lives across the globe. Early diagnosis of the infection is crucial to curb the community spread and enforcing strict quarantine procedure. Real time polymerase chain reaction (RT PCR) based diagnosis is considered as gold standard, however, is resource intensive. We, therefore, developed a PCR based efficient and specific diagnosis method for detection of SARS-CoV-2. We analysed over 100 genomes of SARS-CoV-2 and identified unique consensus sequences for designing PCR primers and evaluated them. Finally, we could optimize the one-step multiplex PCR directly from the extracted RNA and specific primers of envelope protein (E), nucleocapsid protein (N), and membrane protein (M) of SARS-CoV-2. The PCR products were visualized either in agarose gel or in Bioanalyzer. This method showed more than 95% accuracy compared to RT PCR tests, and therefore, is applicable in most of academic as well as pathological laboratory.
In humans and birds, coronaviruses cause deadly diseases of respiratory tracts that can range from mild to lethal [1]. Among humans, common cold with headache if mild illnesses while lethal cases can cause MERS or SARS it emerged as Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS- CoV) in 2002 and 2012 respectively which alternatively caused severe threats to public health [2]. In the late December 2019, another zoonotic form of human coronavirus was emerged in Wuhan, Hubei Province of China, and identified as SARS-CoV-2 that resulted in an outbreak of Coronavirus Disease 2019 (COVID-19) [3]. Since this outbreak, COVID- 19 pandemic infected more than 37 million people globally resulted in death of more than one million people [4]. Recent studies showed that about 30% of infected individuals by SARS-CoV-2 remain asymptomatic [5]. These asymptomatic patients act as carriers which may unknowingly infect a mass of population [6]. Therefore, it became necessary to test as many individuals as possible from the various communities to diagnose for SARS-CoV-2 infection.
COVID 19 can be diagnosed by serological or nucleic acid detection- based assays. Serological assays mainly depend on the detection of antibodies against the spike protein in the serum of infected individuals [7]. Serological assays have some limitations, as very less is known about the nature of immune response to SARS-CoV-2 infection and the scientific basis for durable immunity is not so well developed [8]. Though the assays depending on the detection of antibodies against the viral proteins are useful for serosurveillance, not all are suitable for diagnosis of SARS-CoV-2 due to poor understanding of the antibody responses against SARS-CoV-2 and lack of clarity about the clinical utility of serological testing [9]. Among humans, antibodies usually appear after 5 to 7 days after exposure to the SARS-CoV-2 [10]. During this time, infected individual may continue to spread the infectious viral particles to others and unknowingly infect public [6]. Nucleic acid-based assays are popular choice due to its high accuracy compared to various available serological assays for COVID-19 [11]. Antigen tests are also less sensitive and could be less reliable in case of clinical diagnosis of COVID-19 patients with low viral load [12]. Furthermore, nucleic acid- based assays provide quick results confirmation about infection in comparison to serological tests as the later works on the availability of SARS-CoV-2 protein in infected person.
SARS-CoV-2 is a positive sense single stranded RNA virus having the genome size of about 30 Kb which comprises 11 open reading frames (ORF) [13]. The 5’ of most of ORF encodes a polyprotein which is processed by a virally encoded protease into 15 small peptides, including the RNA dependent RNA polymerase (RdRp) and a non-structural Protein 1 (NSP1). The Envelope (E), Spike (S) and Nucleocapsid (N) genes are encoded by unique independent ORFs [14]. Surface glycoprotein or spike (S) is the longest of these four proteins, it interacts with the host’s angiotensin-converting enzyme 2 (ACE2) receptor and initiates the cascade of reaction that alternatively causes the infection, therefore, it plays a crucial role in rapid transmission from human to human [15]. Nucleoprotein (N) or Nucleocapsid phosphoprotein or interacts with the viral genome and M protein during virion assembly and plays important role in improving the efficiency of sub-genomic viral RNA transcription and viral replication [16]. Small envelope membrane protein (E) like M protein, forms pentameric protein-lipid pores which allow ion-transport and self-assembles inside the host membranes. It also plays a vital role in virus assembly and morphogenesis [17].
Smaller genome size of SARS-CoV-2 makes it prone to accumulate mutations at a much higher rate compared to the organisms having larger genome size. After several rounds of replications in a mass of population, viruses may accumulate a variety of mutations which could result in the loss of infectivity [18]. Single stranded RNA viruses (ss RNA) have higher mutation rates than DNA viruses [19].
Recent advances in polymerase chain reaction (PCR), have allowed scientists for more reliable detection of pathogen for surveillance and identification [20]. PCR requires cycling of temperatures; denaturation at 90 – 95⁰C, annealing at 55 – 60 ⁰C, and extension at 68 – 72 ⁰C to achieve exponential amplification of target DNA using a unique set of oligonucleotides and a thermostable DNA polymerase [21]. The reaction is done in a thermal cycler which works on the principle of Peltier effect [22], the PCR amplicon is mixed with any of the DNA binding fluorescent dye such as ethidium bromide, gel red etc. and amplified DNA is visualized on a UV – transilluminator after its separation on agarose gel. The molecular size of the amplicon is calculated by comparing its position and corresponding band of DNA ladder on agarose gel, DNA ladder contains regularly spaced DNA bands of known intensity and sizes [23].
In RT-PCR (Reverse Transcriptase - polymerase chain reaction) reaction, RNA is first converted to cDNA using reverse transcriptase enzyme and random hexamers or oligo-dT primers. The resultant cDNA is used as template for PCR amplification using gene specific primers. Amplification of specific segment of RNA could be done using RT-PCR, and the amplified PCR products could be visualized on UV- transilluminator [24]. This method of PCR amplification of RNA (through cDNA) has been routinely used in several molecular biology laboratories. Various methods can be used for analysis of DNA fragments size distribution as alternative to commonly used agarose gel electrophoresis which includes chip-based electrophoresis, and capillary electrophoresis [25]. The Bioanalyzer is the first commercially available system which employ chip-based separation technology to separate nucleic acids. The LabChip works on principle of capillary electrophoresis to separates nucleic acid fragments in a chip with microfabricated channels which also automates the detection as well as online data evaluation. This chip based electrophoresis system is widely used for determining the molecular size and quantity of DNA fragments [26-27]. Analysis of PCR products with the bioanalyzer yields several important advantages compared to traditional slab gel electrophoresis as it sizes and quantitates 12 samples on a disposable chip in approximately 30 min. Due to short separation channel and a high electrical field strength the speed of analysis is significantly increased compared to slab gel electrophoresis [27]. This high speed of analysis results in an increased sample throughput. The instrument is equipped with a fluorescence detection system resulting in superior detection sensitivity [27].
In present study, we optimized the multiplex PCR conditions directly from RNA with three SARS-CoV-2 genes namely; E gene, N gene, and M gene corresponding to envelope protein (E), nucleocapsid protein (NC), and membrane protein (M) respectively. Multiplex PCR products can be visualized digitally on computer using Bioanalyzer. This optimized method is helpful detecting the presence of SARS-CoV-2 in one-step directly from RNA. Therefore, this method is cost effective and less time-consuming. It can be easily applied to small molecular biology laboratories having the basal level 2 facility. This method can be used as a toolkit for COVID detection.
Materials and Methods
Collection of samples and Isolation of viral RNA
Naso-pharyngeal swabs were collected and stored in Viral Transport Medium (VTM) as per the standard procedures prescribed by the World Health Organization (WHO) and the Indian Council of Medical Research (ICMR) [28-29]. Testing of samples for COVID-19 were made by following the appropriate ethical guidelines and required consent. Total RNA was isolated from VTM using MagMax RNA isolation kit by following manufacturer’s instructions. RNA was quantified using NanoDropTM spectrophotometer. RNA was either used immediately for PCR based assays or stored at -70°C, until further use.
Single step reverse transcription (RT) and PCR amplification
To do the multiplex PCR, primer sets for three genes were designed from SARS-CoV-2 genome. The detailed list of primers for partial amplification of N, M, and E genes of SARS-CoV-2 are given in Table 1. These primers were synthesized using phosphoramidite method [30]. A primer set was also designed for partial amplification of human beta-actin gene (ACTB) which was used as positive control to check the quality of purified RNA from swab (Table 2). For a single sample, PCR was done in two individual tubes. One tube contained mix of six oligonucleotides called VPM (Viral Primer Mix), and another tube contained mix of two oligonucleotides called CPM (Control Primer Mix). The details of ‘viral primer mix’ is listed in Table 1, and the details of ‘control primer mix’ is listed in Table 2. A positive viral template control (VTC) and a negative no template control (NTC) assay was carried out in each experimental run. For VTC, 1.5 µL of control templates were used, while for NTC, nuclease free water was used instead of template. 3.0 µL of viral RNA was added to a 20 µL reaction mixture containing 2.0 µL of 10x VPM or CPM, 5 µL 4X master mix for one-step PCR (from RNA), and 10.0 µL of nuclease free water. Final concentration of ‘N’ and ‘M’ primers was 250 pmol each, and of ‘E’ primer was 400 pmol respectively. To make easier reaction set-up, 10x stock of VPM was made by adding the 2.5 µL each of N and M primers, and 4.0 µL of E primers from 100 µM stock, and final volume made to 100 µL. During reaction set-up, 2.0 µL from 10x VPM stock was used in a total reaction volume of 20 µL for performing multiplex viral PCR. Similarly, control reaction mix (20 µL) for ACTB was done by adding 2.0 µL from 10x CPM stock. This stock was made by adding 5.0 µL of ACTB 100 µM stock. The final concentration of primer in final PCR reaction mix was 500 pmol. Reactions were carried out in normal thermal cycler. Various permutation and combinations of primer concentration were tried for multiplex PCR and the best results were obtained following thermal cycling conditions: 25⁰ C for 2 min, 53⁰ C for 10 min, 95⁰ C for 2 min, [95⁰ C for 3 sec., 60⁰ C for 30 seconds] X 40 cycles.
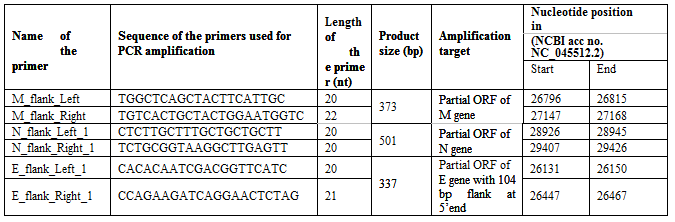
Length of each primers, product size of the amplification, and location of the primers in the genome of SARS-CoV-2 (NC_045512.2) is showed in the table. The table also showed amplification target, i.e., the region of viral genome which was amplified by each primer pair. Mix of all these primers are represented in the following text as “viral primer mix”
Product visualization:
In 10 μL of PCR products 1 μL of gel red DNA visualization dye (Sigma- Aldrich) was added and products were separated on 2% agarose gel (in 1X TAE buffer). 100 kb plus DNA ladder (New England Biolabs) was used to determine size of various amplicons. A gel doc system (BIORAD) was used to visualize the gel.
Data analysis in automated electrophoresis:
Microfluidics-based electrophoresis system namely Bioanalyzer (Agilent 2100) is used for analysis of the PCR products. Optimization of the process were done as per manufacturer’s instruction to achieve faster analysis, less hands-on activities, and analysis report preparation.
Sample preparation and loading PCR products on LabChip (Agilent): Analysis of PCR products was done using an Agilent DNA 1000 kit (Bioanalyzer) using 2100 Expert Software. The instrument uses a 16 well DNA LabChip (Fig. S1), where the PCR products were loaded as per the manufacturer’s instructions.
Development of control plasmids for partial viral genes
To develop the DNA library and positive controls for future experimental validation, Phusion® high-fidelity PCR master mix with HF buffer was used for amplification of partial fragments of N, E, and M genes using cDNA template [31], prepared from purified RNA from SARS-CoV-2 infected person swab. From same cDNA sample human control ACTB gene partial fragment was also amplified. CDNA was synthesized using the RevertAid cDNA synthesis kit by following the manufacturer’s protocol. CDNA was diluted four times in nuclease-free water, and 4 µL of diluent was used as template for PCR synthesis of partial gene fragments of SARS-CoV-2. All the amplicons from viral genes; ‘E’ gene, ‘N’ gene, ‘M’ gene and, human control gene ‘ACTB’ were gel purified using QIAquick gel extraction kit and cloned in Zero blunt TOPO PCR cloning kit using the manufacturer’s protocol. A molar vector to insert ratio of 1:5 was used for ligation, assembled products were transformed in chemically competent DH5ά E. coli cells. Transformed colonies were screened on LB agar plates containing 50 µg/mL kanamycin [32]. Clones were initially confirmed for their integration in vector using M13 forward and reverse primers, and results were analysed by comparing the molecular sizes of respective amplicon with 100 kb plus DNA ladder.
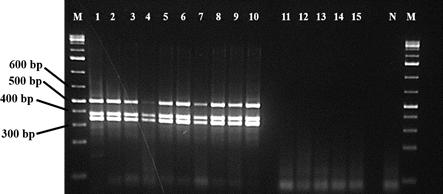
All the three partial viral genes were amplified from cDNA individually (Fig. 6) and cloned in Zero blunt TOPO PCR cloning kit. Successful cloning was confirmed by sanger sequencing of cloned fragments. Further confirmation was done by restriction digestion using EcoR1 (Fig. S2), which released the product of respective sizes for each of the partial gene. The plasmids were sequenced using M13 forward and M 13 reverse primers in 3730 DNA analyzer (ThermoFischer Scientific) using Big-dye terminator kit. The sequences were compared with the original viral nucleic acid to confirm accuracy.
Results and Discussion
Specificity of the primers
Over 100 genomes of the SARS-CoV-2 were retrieved from NCBI database and subjected to multiple sequence alignment (MSA) to know how much variations are available among the genomes. Based on the alignments, we identified the conserved consensus sequences from various genes, and used as primers for amplification of partial N, E, and M genes of SARS-CoV-2. Detailed list of primers and their respective amplicon sizes are mentioned in the table 1. The nucleotide sequences of N, M, and E gene along with 200 bp flanking regions from both 3’ and 5’ ends were subjected to multiple sequence alignment studies.
Total RNA from SARS-CoV-2 infected person was purified using MagMax RNA extraction kit. The quality and quantity of RNA was evaluated through nanodrop spectrophotometer by measuring the 260/280 ratio. The RNA sample having this ratio in between 1.9 to 2.1 was used for the study.
One-step multiplex PCR
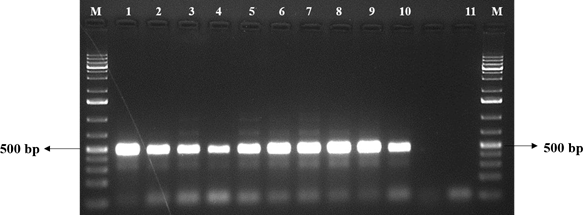
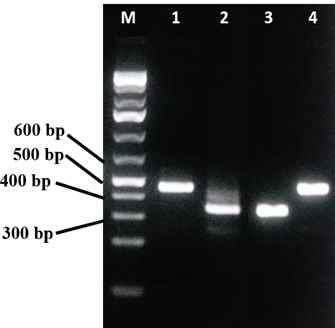
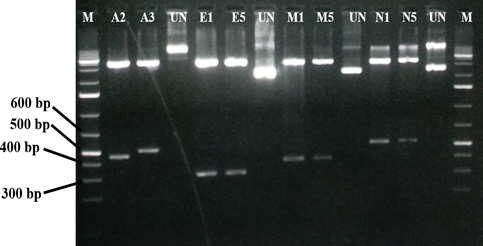
We did the normal PCR of human control ACTB gene (436 bp) to check the quality of purified RNA (Fig. 1). It helped in understanding of all the steps in RNA isolation and protocol worked properly. After confirming the sample quality, total RNA was used as template for both viral multiplex and human control gene PCR. The Multiplex SARS-CoV-2 PCR product was separated on 2 % agarose gel, three distinct bands of respective sizes; 373 bp for M gene, 501 bp for N gene, and 337 bp for E gene were seen on agarose gel (Figure. 2).
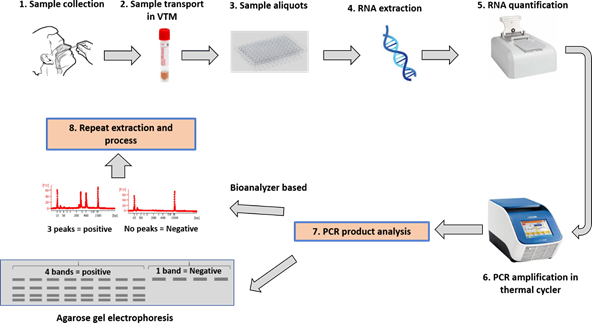
1 sample collection followed by (2, 3, & 4) transport, aliquots and extraction. Extracted samples are then (5) quantified and good quality samples are (6) amplified in thermal cycler as describe above. Amplified PCR products are then (7) analyzed by any one or both methods described agarose gel based as well as bioanalyzer based. Expected results not obtained or any error occurred then whole (8) process can be repeated
Bioanalyzer based detection:
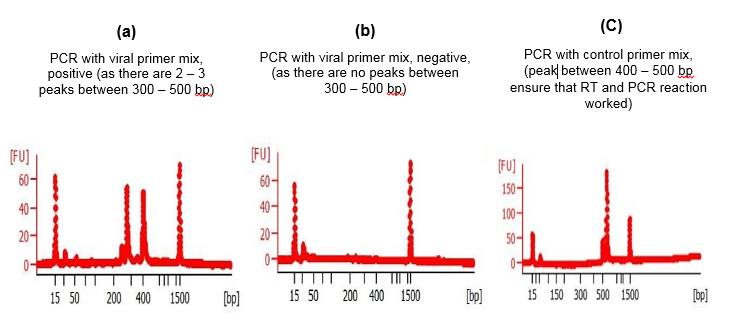
Analysis using digitalized platform of Bioanalyzer generated high-quality digital gel images on computer and showed the respective amplicon sizes [33]. It also showed intensity-based quantity of various viral gene bands in multiplex PCR (Fig. 2, 4). The Labchip accommodates wells for samples, gel and an external standard (ladder). To create interconnected networks among the wells, micro-channels were made in glass. A sieving polymer was filled in the micro-channels during chip preparation. Once the wells and channels are filled, the chip becomes an integrated electrical circuit. The 16-pin electrodes of the cartridge are arranged so that they fit into the wells of the chip. Each electrode is connected to an independent power supply that provides maximum control and flexibility. A voltage gradient like slab gel electrophoresis was used to drive DNA and RNA biomolecules electrophoretically. Molecules got separated based on size because of the presence of a sieving polymer matrix and constant mass- to-charge ratio [34-35]. The detection is automatically performed by laser induced fluorescence (LIF) detection. Data is translated into gel-like images (bands) and electropherograms (peaks) where the X axis represent size and Y axis represents quantity of the PCR product. This principle was used to separate the SARS-CoV-2 multiplex PCR products using this digitalized platform. A standard curve of migration time versus molecule size is plotted with the help of a ladder that contains components of known sizes. Analysis of the samples containing SARS-CoV-2 virus (positive) produced 3 ‘viral PCR peaks’ between 300 – 500 bp (Fig. 4a). Whereas the samples which does not contain the virus (negative) does not produced any peak between 300 – 500 bp region (Figure. 4b). The PCR products containing ‘control primer mix’ produced only single peak within 400 – 450 bp in both positive and negative (Fig. 4c). Appearance of this peak ensured that RT and PCR reaction has worked properly in a single reaction tube. The general diagram showing the chip structure and its micro-fluidic design is shown (Fig.S1). the electropherogram produced after the digitalized gel electrophoresis in bioanalyzer DNA chip device are also shown (Figure 4, 5).
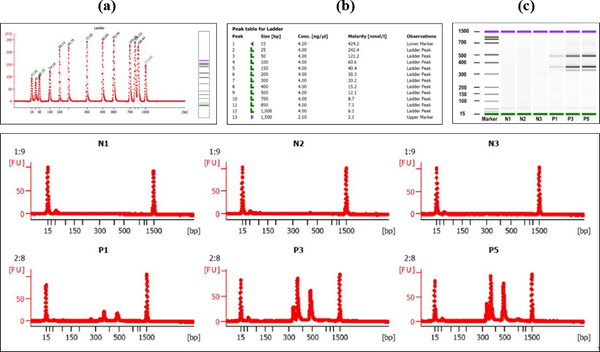
Partial DNA region of envelope protein (E), nucleocapsid protein (N), and membrane protein (M) of SARS-CoV-2 were cloned in plasmid vector used as positive control in PCR analysis of the patient samples. These control DNA were used to validate the reaction condition and quality assay of the primer-mix used in the study (Figure. 6). In the quality assay the primer pair or mix is expected to generate PCR products of 436 bp for actinB gene, 337 bp for E gene, 373 BP for M gene, 501 bp for N gene (Figure. 6).
As there has been a sudden increase in the COVID-19 cases worldwide it becomes necessary to have efficient testing facility to overcome and control the pandemic situation. Various alternative methods and assays for RT PCR are now commercially available in the market but not all are that efficient in detection of viral infection. Though antibody testing is a rapid method for detection but have certain limitations as it detects the antibodies a week period after the infection and till that time, infected person can spread infection to others. As per several reports, antigen test is also giving about 50-60% accurate results and not reliable for sample with low viral load. As there are challenges in developing specific antibodies for COVID-19, antigen test becomes less favorite for testing. Though RT PCR known to be gold standard for testing COVID-19 detection, its sensitivity gets affected by low viral load into the patient sample [36]. This assay development involves huge investment as it requires expensive instruments and skilled person to operate so increases overall costing of test, small laboratories and institutions cannot afford it. By contrast, sensitivity of our multiplex PCR assay did not have any issue due to low viral load and found more sensitive than standard RT-PCR. Developed method to be useful in diagnosis of SARS-CoV-2 in small laboratories or residential on a low cost. It can be utilized for regular testing of employees working, or people residing in the small housing society. This method can do low throughput in short time, which reduces the cost as well. The method can be further improvised to perform high throughput and save more time and cost.
Supplementary Material (ESM)
The current study was funded by Reliance Industries Limited
The authors declare they have no conflict of interest.
The authors thank Dr. Ipsita Dutta, Dr. Yash Gupte, Dr. Smita Patil for their valuable support and Reliance Life Science for providing infrastructural and logistic support during this study. We also thank to RIL – IP team for review of the manuscript and providing approval for publication.
Compliance with ethical standards
Ethical approval This article does not contain any studies with human participants or animals performed by any of the authors.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
