
case Report | DOI: https://doi.org/10.31579/2690-4861/620
¹Endocrinology and Metabolism Resident at Mackenzie University Hospital.
²Internal Medicine Resident at Mackenzie University Hospital.
³Head of the Endocrinology and Metabolism Department at Mackenzie University Hospital.
*Corresponding Author: Heloisa Lima, Endocrinology and Metabolism Resident at Mackenzie University Hospital.
*Corresponding Author: Heloisa Lima, Endocrinology and Metabolism Resident at Mackenzie University Hospital.
Citation: Heloisa Lima, Emanuelle Leonel, Eduarda Menestrina, Caio Hayashi, Bruno Kliemann, et al, (2025), Not So Fahr: A Better Look at Endocrine Abdnormalities in Rare Neurological Conditions, International Journal of Clinical Case Reports and Reviews, 22(5); DOI:10.31579/2690-4861/620
Copyright: © 2025, Heloisa Lima. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 14 November 2024 | Accepted: 26 November 2024 | Published: 29 January 2025
Keywords: hypocalcemia; hypoparathyroidism; Fahr’s syndrome; basal ganglia calcification
Fahr’s syndrome is a rare neurological condition characterized by calcification of the central nervous system. Endocrine diseases related to calcium metabolism are known to cause this pathology. In the literature, for instance, etiological descriptions of this syndrome are found in hypoparathyroidism and pseudohypoparathyroidism; however, its prevalence remains unknown.
This syndrome is characterized by progressive cognitive deterioration, movement disorders, neuropsychiatric alterations, and bilateral calcifications in the central nervous system, typically manifesting between the third and fourth decades of life. As mentioned, it can be secondary to other diseases, and the early and correct treatment of those can prevent progression to Fahr’s syndrome.
The objective of this case series, along with a literature review, is to highlight this differential diagnosis. In “Not so Fahr,” we describe the cases of two patients who were simultaneously admitted to Mackenzie University Hospital (Curitiba, Brazil), presenting with similar clinical and imaging findings, the same syndromic diagnosis, but different etiological diagnoses.
Fahr's syndrome, first described by Karl Fahr in 1930, is a rare neurological condition characterized by calcification of the central nervous system, presenting with variable clinical manifestations and independent of its etiology [1]. This condition follows an autosomal dominant inheritance pattern, is idiopathic, and secondary to an underlying disease. It differs from its primary form, known as Fahr's disease, which is generally associated with familial forms [1,2,3].
In this syndrome, the calcifications consist of calcium salts, iron, phosphate, and other minerals that accumulate and deposit within and around the blood vessel walls of the basal ganglia, as well as in the dentate nucleus, cerebellum, hippocampus, thalamus, and other regions [1,2]. Consequently, neurological manifestations are noted, such as progressive cognitive deterioration, movement disorders, neuropsychiatric alterations, and bilateral calcifications in the central nervous system [2,3]. As such, it can be an important factor to impact the quality of life.
In cases of endocrine disorders, this syndrome is related to calcium metabolism, especially in hypoparathyroidism and pseudohypoparathyroidism, being one of the manifestations of chronic hypocalcemia. It is more common in patients who have been untreated for a sufficiently long period [1,2,3]. Early treatment of parathyroid disorders and electrolyte imbalances prevents progression to irreversible complications and generally controls neurological manifestations.
In this article, the Endocrinology Department of the Evangelical Mackenzie Hospital presents two clinical cases of strio-pallido-dentate calcifications related to calcium metabolism disorders, where two patients exhibited similar laboratory findings and syndromic diagnoses but different etiological diagnoses. Both cases culminated in a common condition: Fahr's syndrome. These patients were recently admitted for initially neurological issues and were subsequently monitored by the authors of this article. A brief literature review is also included in this work.
Clinical Case 1: Fahr's Syndrome in Pseudohypoparathyroidism
A female patient, KSC, currently 18 years old. At age 9, she began her diagnostic investigation due to episodes of seizures, which led to her hospitalization at the time, where hypocalcemia was detected. Since then, numerous diagnostic tests have been conducted, revealing persistently elevated PTH levels, yet paradoxically showing hypocalcemia and hyperphosphatemia (Table 1). On clinical assessment, the patient does not have stigmata on physical examination (Figure 1), has a height of 1.65m—within her family growth channel—and maintains average academic performance (having repeated one year of elementary school).
| Exam Date | PTH (pg/mL) | Calcium* (mg/dL) | Phosphorus (mg/dL) | Vitamin D (ng/mL) |
| 16/07/2014 | - | 5,10 | 6,4 | - |
| 16/10/2014 | 524 | 5,6 | 9,3 | 62,6 |
| 01/09/2021 | 90 | 9,9 | 3,9 | 18,2 |
| 22/03/2022 | 422 | 9,5 | 4 | 28,6 |
| 24/04/2022 | 513 | 6,9 | 4,9 | 28,2 |
| 25/10/2022 | 505 | 6,9 | 4,8 | 22 |
| 07/01/2023 | 499 | 6,1 | 4,9 | - |
| 13/07/2023 | - | 6,6 | 5,2 | 26 |
| 25/11/2023 | - | 9,05 | 4,83 | - |
Table 1: Laboratory tests of patient KSC. Reference values from the laboratory: PTH 12-88 pg/mL, Calcium 8.8-10.6 mg/dL, Phosphorus 2.5-4.5 mg/dL, Vitamin D (25-OH-Vitamin D) greater than 20 ng/mL. *Calcium values corrected for albumin.
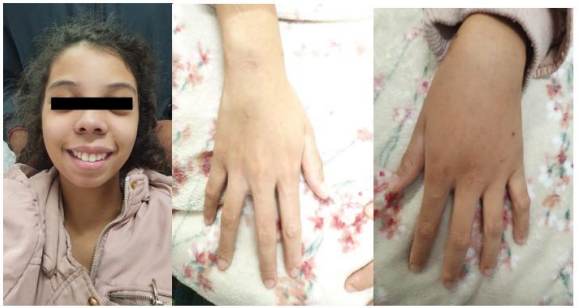
Figure 1: Ectoscopy of patient KSC at 17 years old.
Due to symptomatic hypocalcemia, with muscle spasms and seizures, the patient undergoes daily supplementation of calcium carbonate and vitamin D (1250 mg/400 IU in 7 daily tablets), along with calcitriol (0.25 mg in 6 daily tablets) and hydrochlorothiazide (25 mg per day). Despite this, she still occasionally experiences hypocalcemia and elevated PTH levels. Given the absence of physical stigmata and the laboratory findings, the patient was diagnosed with pseudohypoparathyroidism type 1B.
In May 2024, the patient was re-hospitalized at Evangelical Mackenzie Hospital due to seizures. Upon admission, laboratory tests revealed a corrected serum calcium level of 7.8 mg/dL, phosphorus at 4.05 mg/dL, and magnesium at 1.65 mg/dL. A cranial CT scan was performed, showing symmetric calcifications in the basal ganglia, thalami, and diffusely in supratentorial subcortical regions (Figure 2). Thus, a clinical and radiological diagnosis of Fahr's syndrome was confirmed.

Figure 2: Cranial CT scan of patient KSC, showing symmetric calcifications in the basal ganglia, thalami, and diffusely in supratentorial subcortical regions.
The patient showed good control of seizures with the use of the following anticonvulsants: valproic acid, carbamazepine, and phenytoin in combination, as directed by the neurology department of the hospital. She was discharged after 22 days of hospitalization, continuing with the prescribed supplementation of calcium, calcitriol, and cholecalciferol for outpatient follow-up with endocrinology, in addition to treatment for seizures.
Clinical Case 2: Fahr's Syndrome in Post-Surgical Hypoparathyroidism
A female patient, A.V.P.S, 52 years old, underwent total thyroidectomy for plunging goiter at age 30. She was admitted to the emergency room of Mackenzie University Hospital with her first episode of seizures precipitated by a urinary tract infection.
Upon admission, the patient had a surgical scar on her neck and a positive Chvostek sign (Figure 4), making the diagnosis of hypoparathyroidism highly suspect, which was later confirmed by laboratory tests (Table 2).

Figure 3: Patient with dry and rough skin characteristics. Brittle hair, alopecia – Chvostek sign recorded in the image (video capture).
The patient presented the following laboratory results upon admission:
| Exam date | PTH (pg/mL) | Calcium* (mg/dL) | Phosphorus (mg/dL) | Vitamin D |
| 24/05/2024 | <1> | 6,82 | 5,48 | 16.4 |
Table 2: Laboratory tests of patient A.V.P.S. Reference values from the laboratory: PTH 12-88 pg/mL, Calcium 8.8-10.6 mg/dL, Phosphorus 2.5-4.5 mg/dL, Vitamin D (25-OH-Vitamin D) greater than 20 ng/mL. *Calcium values corrected for albumin.
Investigations were conducted with an electroencephalogram, fundoscopy, and cranial computed tomography. The electroencephalogram revealed rare epileptiform activity localized in the left frontotemporal region. The fundoscopy showed no alterations. Finally, the computed tomography (Figure 5) confirmed the diagnosis of Fahr's syndrome secondary to hypoparathyroidism.

Figure 4: Cranial CT scan of patient A.V.P.S, showing symmetric intracranial calcifications.
The patient was treated with calcitriol and intravenous infusions of calcium gluconate until normalization of calcium levels. Similar to patient 1, she was discharged with elemental calcium administered at a dose of 1-2 g/day and calcitriol, showing significant clinical improvement.
Initially described by Karl Fahr in 1930, Fahr's syndrome is recognized as a condition characterized by neuropsychiatric and behavioral changes secondary to calcifications in the central nervous system. As a classification, the syndrome differs from Fahr's disease, as the latter has a primary cause, which can be familial with autosomal dominant inheritance or sporadic in nature [1,2].
Fahr's disease is characterized by progressive cognitive deterioration, movement disorders, neuropsychiatric alterations, and bilateral calcifications in the central nervous system, typically manifesting between the third and fourth decades of life. Additionally, in Fahr's disease, it is crucial to exclude secondary factors that may lead to such findings, such as biochemical alterations, infections, or trauma. In contrast, Fahr's syndrome encompasses a broad range of diseases that cause secondary calcifications in the central nervous system but may present with a clinical picture similar to that of Fahr's disease [1,2].
Calcium metabolism disorders are directly related to levels of vitamin D and PTH, as well as fibroblast growth factor 23 (FGF23), phosphate, and calcium itself. Hypocalcemia, on the other hand, is more closely associated with the first two factors [4,5]. It is defined based on low levels of total serum calcium and ionized calcium— it is noteworthy that serum calcium levels are directly influenced by albumin levels, and corrected serum calcium should always be evaluated [4,5,6].
Among the most important causes of hypocalcemia are parathyroid disorders, particularly hypoparathyroidism and pseudohypoparathyroidism. These are also common causes of neurological disorders, which may manifest as paresthesias, tetany, papilledema (pseudotumor cerebri), extrapyramidal signs, mental retardation, personality changes, depression, and seizures [4,5,6].
In general, the diagnosis arises from neurological complaints, as hypocalcemia increases central and peripheral neuronal irritability, and seizures may be the initial manifestation, especially in very young or elderly patients [4,5]. By lowering the excitatory threshold of preexisting epilepsy, seizures are indistinguishable from those that occur in the presence of normocalcemia and can manifest in various forms [4,5,6].
Regarding hypoparathyroidism, postoperative etiology is its most frequent cause, but there is a range of differential etiologies, and its most important biochemical changes are hypocalcemia and hyperphosphatemia, in the presence of normal renal function. [5]
Serum calcium concentrations vary from 6-7 mg/dL to values close to normal, depending on the severity of the disease. Phosphorus is generally elevated, between 6-7 mg/dL. Immunoreactive PTH is low or undetectable, except in cases of PTH resistance, where levels are elevated. Magnesium levels may be decreased due to reduced intestinal absorption and tubular reabsorption of this ion [4,5,6].
The incidence of hypoparathyroidism is often less than 5%, generally ranging from 1% to 2% of parathyroid surgeries [6]. Since cataracts can be an associated manifestation, fundoscopy should be part of the initial evaluation of these patients.
Success in treating hypocalcemia (which is done with calcitriol and calcium supplementation) often prevents seizures, potentially reducing or ceasing the need for anticonvulsants. In cases of postoperative hypoparathyroidism, calcifications are observed on average 17 years after surgery [6].
Hypoparathyroidism with neurological manifestations diagnosed many years after thyroid surgery is considered a rarity [7], as postoperative follow-up allows for the prompt recognition of hypocalcemia, and its correction seems to prevent calcifications. It is known that neurological symptoms can be precipitated or worsened by hyperventilation, intense exercise, withdrawal of thyroid medication, infection, metabolic acidosis, uremia, and phenytoin. During the premenstrual period and in pregnancy, symptoms may worsen due to salt retention. Additionally, electroencephalogram (EEG) changes persist after the correction of hypocalcemia, despite the decrease in the number of seizures. [6,7].
Another etiological diagnosis within the spectrum of hypocalcemia is pseudohypoparathyroidism (PHP). The pathophysiology of this disease consists of peripheral resistance to PTH, which usually occurs in bones and kidneys, due to a mutation in the hormonal receptor that leads to a defect in the activation cascade by the GNAS subunit – inherited in an autosomal dominant manner [7]. Thus, the laboratory alterations frequently found in this case are elevated PTH levels, hypocalcemia, and hyperphosphatemia [8,9].
The prevalence of PHP remains uncertain due to the difficulty of laboratory confirmation at the molecular analysis level. It is estimated to be a rare disease – between 2000 and 2016, fewer than 60 cases were reported worldwide [9].
Pseudohypoparathyroidism is distinguished into different subtypes, each with different clinical manifestations. In PHP type 1A, the classic form of the disease, there is a reduction in GNAS levels, leading to peripheral resistance to PTH, but there may also be resistance to TSH [8,9]. Its clinical presentation usually occurs in childhood and is related to Albright's Hereditary Osteodystrophy – physical examination findings include a rounded face, short stature, brachydactyly, subcutaneous calcifications, obesity, and cognitive impairment. When present, cognitive delay correlates with the severity of the disease; however, up to 30% of patients with PHP type 1A do not exhibit this finding [8,9,10].
PHP type 1C is similar to 1A regarding physical examination findings and osteodystrophy; however, it presents normal levels of the GNAS subunit. Some authors consider it a subtype of 1A [9,10].
PHP type 1B is distinguished from the first two by the isolated resistance of PTH at the renal level. As a result, there are normal GNAS levels and an absence of the findings of Albright's Hereditary Osteodystrophy; however, in some cases, patients may present some of the findings, especially brachydactyly. In these patients, peripheral resistance to other hormones, such as TSH, may also be present. This subtype of pseudohypoparathyroidism can be inherited maternally but can also occur sporadicall [8,9].
PHP type 2 is characterized by increased levels of cAMP in response to exogenous PTH but without compatible phosphaturic response. The pathophysiology remains uncertain and may be a secondary effect of vitamin D deficiency [8,9].
Finally, pseudo-pseudohypoparathyroidism consists of a reduction in the GNAS subunit, with phenotypic changes of Albright's Osteodystrophy but without resistance to PTH. Thus, it has no laboratory alterations – normal levels of calcium, phosphorus, and PTH [7,9,10].
The clinical manifestations of pseudohypoparathyroidism include, in addition to those already mentioned, endocrine-metabolic alterations (elevated TSH, hypogonadism, GH deficiency), neurological issues (auditory alteration, carpal tunnel syndrome, craniosynostosis), mineralization defects (calcification of the central nervous system/Fahr syndrome, oligodontia/hypodontia, cataract), among others. Diagnosis should be suspected based on clinical and laboratory findings [9,10]. Genetic evaluation includes DNA sequencing, methylation, analysis of the GNAS locus, and should be indicated for patients with a high suspicion [7].
The treatment of PHP should preferably be conducted by a multidisciplinary team, including genetic counseling. It is recommended to measure calcium, phosphorus, and PTH levels every six months before the start of treatment, with more frequent evaluations of calcium levels in certain cases (growth phases, pregnancy, lactation, acute diseases) [9,10]. Treatment consists of vitamin D replacement (preferably calcitriol), and calcium supplementation should be based on dietary levels. The goal of treatment is to maintain normocalcemia and normophosphatemia while avoiding hypercalciuria. There is no recommendation for the replacement of PTH or its analogs [8,9,10].
The prevalence of calcifications in the basal ganglia in endocrine disorders is still unknown; however, it is a "not so Fahr" condition within the hospital environment and should therefore be included in the differential diagnosis by the clinician.
Early treatment of parathyroid disorders and electrolyte imbalances prevents progression to irreversible complications and generally controls neurological manifestations.
Understanding the pathophysiology, therapeutic management, and prognosis of both etiologies is essential for therapeutic management, focusing on restoring calcium and phosphorus levels near to normal, preventing complications, and ensuring multidisciplinary follow-up.
Conflicts of Interest
*The authors of this work have no conflicts of interest.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
