North manchester general hospital, delaunays roadmanchester, m8 5rb. United Kingdom.
*Corresponding Author:
Anthony Kodzo-Grey Venyo, North manchester general hospital, delaunays roadmanchester, m8 5rb. United Kingdom.
Citation: Grey Venyo AK, (2024), Neuro-Endocrine Tumours of Testis: a Review and Update, Clinical Research and Clinical Trials, 9(1); DOI:10.31579/2693-4779/164
Neuroendocrine tumours more frequently arise from the embryonic gut. Neuroendocrine tumour of the testis could occur as a primary tumour or as metastases with the primary tumour originating elsewhere within the body. Primary neuroendocrine tumours of the testis are not common tumours. To the knowledge of the author, less than 200 cases of Neuroendocrine tumours of the testis had been published up to date. The cell of origin of neuroendocrine tumour is not definitely known. It has been iterated that neuroendocrine tumour commonly arises from intestinal and respiratory epithelium. Neuroendocrine tumour of the testis which is an uncommon tumour of the testis has been documented to account for less than 1% of all tumours of the testis. To the knowledge of the author, less than two hundred (<200) cases of neuro-endocrine tumour of the testis had been reported in the literature. Neuroendocrine tumour of the testis can afflict a child or an adult. Majority of patients who are afflicted by neuroendocrine tumour of testis manifest with unilateral painless testicular mass. It has been documented that 16% of patients who have neuroendocrine tumour of the testis manifest with symptoms of neuroendocrine tumour syndrome. and about eleven percent of primary testicular neuroendocrine tumours had been documented to be associated with metastasis. Neuroendocrine tumours of the testis do occur as a primary testicular neuroendocrine tumour of testis; or metastatic neuroendocrine tumour to the testis. Primary neuroendocrine tumour of the testis does get to be further sub-divided into: (a) primary pure testicular neuroendocrine tumour; and (b) testicular neuroendocrine tumour which is associated with teratoma or dermoid/epidermoid cysts. It has been pointed out that once a testicular neuroendocrine tumour has been diagnosed a metastatic neuroendocrine tumour to the testis needs be excluded. It has been pointed out that the presence of teratoma elements in the testicular tumour sufficiently excludes the primary site outside the testicles. It is therefore important to submit the tumour mass entirely and to look for any additional lineage of differentiation. It has been documented that twenty-five percent of primary neuroendocrine tumours of testis had tended to be associated with teratoma. Histogenesis of pure neuroendocrine tumour of the testis, has up to date remained unclear. Two postulates had been promulgated regarding the mode of origin of neuroendocrine tumour of the testis including: (a) the tumours are derived from teratoma (germ cell origin) or (b) the tumours do arise within argentaffin cells located in crypts of Lieberkühn. The later are also stated to present within the gastrointestinal tract and bronchial mucosa. Nevertheless, in a report of a study which had been related to three pure testicular neuroendocrine tumours had indicated that pure neuroendocrine tumour of testis, might have different genetic background other than germ cell tumour. Based upon histopathology examination, neuroendocrine tumour has been documented to be typified by presence of nests of small round cells with uniform nuclei forming small acini and rosettes or sheets. The cells of neurogenic tumour of testis do have eosinophilic granules within the cytoplasm and granular chromatin within the nuclei. The neuroendocrine tumour of testis cells, do exhibit positive immunohistochemistry staining for neuroendocrine markers like chromogranin and synaptophysin. Diagnosis of neuroendocrine tumour of testis is established based upon the histopathology and immunohistochemistry staining examination features of the tumour. It has been iterated that radical orchidectomy is the treatment of choice; nevertheless, it has been pointed out that the need for adjuvant therapy depending upon histological grading has remained not clarified. It has also been iterated that Octreotide analogue could improve the neuroendocrine (carcinoid) syndrome as well as stabilize the tumour. Other iterations that had been made regarding neuroendocrine tumour of testis include:
The rarity of neuroendocrine tumour of testis is a limiting factor for the understanding of the biological behaviour of the tumour upon which a modern classification and prediction of prognosis of the tumour could be reached.
Localized neuroendocrine tumour of testis does have an excellent prognosis following the undertaking of surgery
Neuroendocrine tumour of testis could develop metastasis 7 years pursuant to the initial treatment. Few cases had reported metastases 5 years and 19 years subsequently.
Fifty percent (50%) of neuroendocrine tumours of testis associated with carcinoid syndrome metastasized and the larger tumour, then the higher risk for the subsequent development of metastasis.
The association with teratoma and neuroendocrine tumour of testis is stated to has a prognostic value depending upon the age; and post-pubertal teratomatous neuroendocrine has advert prognosis.
In view of the fact that a metastatic potential exists, long-term follow-up is indicated.
Regular clinical examination, urinary 5-HIAA, abdominal CT scan, and gastrointestinal contrast could be undertaken for follow-up assessments of patients who have undergone treatment for neuroendocrine tumour of testis.
Primary testicular neuroendocrine tumour of testis is not common.
It is pivotal to submit the entire gross testicular tumour specimen for histopathology examination to exclude existence of other germ cell elements.
It is pertinent for all patients who have undergone treatment for neuroendocrine tumour of testis to be followed-up for a long-time to enable prompt confirmation of local recurrence or metastasis which could occur early or very late after many years.
Introduction
It has been iterated that testicular adrenal rest tumours (TARTs) are benign intratesticular masses which do occur in male patients who have congenital adrenal hyperplasia (CAH), with more than 90% of adrenal rest tumours that are caused by a deficiency of 21-α-hydroxylase.[1] [2] [3] TARTs had been stated to originate from aberrant adrenal cells within the testes and TARTS could impair both spermatogenesis and endocrine testicular function. [3] Studies had suggested that TARTs might be present in childhood, with an increasing prevalence of TARTS after the onset of puberty.[4] [5] In view of this it has been advised that the early detection of TARTs is necessary in order to preserve testicular function, and scrotal baseline ultrasound (BUS) scan screening had been recommended beginning within early childhood. It had also been iterated that even though many reports of TART ultrasound scan features had been reported previously, the reports had been limited by small numbers of reported cases [6] [7] [8] or the old age of the patients. [4] [9] [10] One of the most important and frequently detected complications in males with congenital adrenal hyperplasia (CAH) is the development of testicular adrenal rest tumours (TARTs). TARTs were first reported in 1940. [4] [11] Since that time, testicular tumours have been described in several publications (mainly case reports). However, several complications can develop in adult patients with CAH, and in the recent years, it became clear that some of patients could have been diagnosed in childhood. Adrenal rest tumours of the testis are frequently detected in adult males with CAH, with the prevalence up to 94%. [12] TARTs in children have mostly been presented as case reports in the literature, [13]. [14] [15] and only a limited number of studies have described the prevalence of TARTs in large populations of children and adults. [7] [16] [17] [18] [19] [20]. Avila et al. [7] reported that the prevalence of TARTs in male patients with CAH between 6 and 31 years of age was 21%, with the youngest case being 6·2 years of age. Vanzulli et al. [16] reported the prevalence of TARTs in 30 male patients with CAH, ageing between 9 and 32 years, was 27%, and the prevalence of TARTs in patients with CAH < 18 years of age was 29%. Importantly, these studies did not focus on children. Prevalence of TARTs was reported as 24% among CAH patients between 2 and 18 years of age in the most comprehensive study conducted on children, and inhibin-assessed gonadal function was normal in children with TARTs [20] Preservation of gonadal functions is associated with tumour size; however, gonadal function of patients with TARTs might deteriorate during adulthood. Moreover, tumour sizes might be increased during the follow-ups, and the increase might subsequently lead to impairment of gonadal function. Therefore, long-term follow-ups are important in patients who were diagnosed during the childhood. The ensuing article on neuroendocrine tumour of testis is divided into two parts: (A) Overview which has discussed miscellaneous general aspects related to neuroendocrine tumour of testis and (B) which includes Miscellaneous narrations and discussions from some case reports, cases series and some studies related to neuroendocrine tumours of testis.
Aims
To review and update the literature on neuroendocrine tumours of the testis.
Methods
Internet data bases were searched including: Google, Google Scholar, PUBMED and Yahoo. The search words that were used included: Neuroendocrine tumours of the testis; Testicular neuroendocrine tumour; Carcinoid tumour of testis; Testicular carcinoid tumour. One hundred and fifty-four (154) references were identified which were used to write the article which has been divided into two parts: (A) Overview which has discussed miscellaneous general aspects related to neuroendocrine tumour of testis and (B) which includes Miscellaneous narrations and discussions from some case reports, cases series and some studies related to neuroendocrine tumours of testis.
Results
A] OVERVIEW
Definition / general statement
Adrenal rest tumour of the testis is associated with congenital adrenal hyperplasia and it is characterized by benign biological behaviour and a typical hormone sensitivity [21]
Terminology
Other terminologies that had been used for adrenal rest tumours of the testis as well as statements that had been made related to the tumour include: [21
Testicular tumours of Adrenogenital syndrome (TTAGS) [21
Adrenal rest tumours of the testis had also been referred to as Testicular adrenal rest tumours (TARTs) [21]
Biologically, adrenal rest tumours of the testis represent hyperplasia of aberrant rests rather than a true neoplastic tumour [21
Epidemiology
It has been pointed out that adrenal rest tumours of the testis can manifest at any age but they commonly manifest in pubertal and post-pubertal males, whose mean age is about 15 years [21]
It has been iterated that the incidence and prevalence of adrenal rest tumour of the testis does vary based upon the age of the patient age and the prevalence of the tumour increases with age, as well as the prevalence of the tumour depends upon geography as follows: [21]
It has been stated that adrenal rest tumour is often clinically undetected [16] [21]
It has also been documented that adrenal rest tumour of testis has been detected at autopsy in 3 out of 7 patients who have congenital adrenal hyperplasia (CAH) who were less than 8 weeks old [21] [22]
It had been reported that there was a 24% prevalence in 34 children who had CAH and whose ages had ranged between 2 years and 18 years. [20]
It has been documented that adrenal rest tumour of testis is associated with congenital adrenal hyperplasia - 21 hydroxylase deficiency [23] [24] and that 27% to- 47% of the patients develop testicular adrenal rest tumours [12] [25], [26]
Other conditions which had been documented to be associated with testicular adrenal rest tumours include Nelson syndrome, Cushing disease, and Addison disease [21].
Sites
It has been documented that adrenal rest tumours normally tend to be found in descended testis and adjacent tissues. [21
Aetiology and Pathophysiology:
Summating iterations that had been made about the aetiology and pathogenesis of adrenal rest tumour include: [21]
Development and pathogenesis
The development of TTAGS is dependent upon the ensuing factors: [21]
Presence of adrenal rests within and around the testis
Abnormal growth is stimulated by excess ACTH or Angiotensin II levels.
Duration and concentration of exposure to growth stimulating hormones
Aberrant adrenal rests in and around the testis had been demonstrated at autopsy in about 7.5% of infants without any pathological significance [27] and in 15% of healthy neonates [28], [29]
It has been stated that probably adrenal rest tumour of the testis had been under reported due to difficulty in identifying adrenal rests. [21]
It has been iterated that adrenal rest tumours of testis are more commonly found in patients who have congenital adrenal hyperplasia who are noncompliant with their treatment. [21]
It has been documented that the Tumour's adrenal origin and functional status had been inferred from in vitro and in vivo studies including: [21]
Adrenal specific enzyme CYP11B1 (11b-hydroxylase) present within tumour tissue of CAH patient who has TTAGS [30]
Adrenal specific steroids tend to be present [31]
Adrenal specific 11b-hydroxylated steroids tend to be found present within blood from gonadal veins of patients who have adrenal rest tumours of testis. [21] [31] [32] [33]
It has been pointed out that in patients who have adrenal rest tumours of testis, mRNA expression seen of adrenal specific enzymes CYP11B1 is expressed by zona fasciculate anzona reticularis and CYP11B2 is expressed by zona glomerulosa), as well as of ACTH and angiotensin II (AII) receptors [34]
It has however been stated that the persistence of tumours despite ACTH suppression indicates the role that is played by other factors. [21]
It has been reported that TTAGS had also been reported in patients with adequately controlled hormone levels [25]
It has been stated that angiotensin II might have role to play in the growth of TTAGS [35]
It has been stated that TTAGS had not been reported in late onset CAH without clearly elevated ACTH or Angiotensin II levels. [21]
It had also been stated that LH might have a role to play in the development and growth of TTAGS. [36]
Effects of TTAGS:
It has been iterated that mechanical effects of TTAGs include: the size and duration dependent blockage of rete testis with resulting atrophy of seminiferous tubules. [21]
It had been stated that the paracrine effects of TTAGS include: steroids that are produced by the tumour cells might be toxic to Leydig cells or germ cells [21] [37]
It has been documented that the irreversible end stage of longstanding TART is tubular hyalinization with obstruction of the lumen and complete loss of germ cells and Sertoli cells and that this differs from ischemic hyalinization by relative preservation of Interstitial Leydig cells. [21]
Clinical features
The clinical features of adrenal rest tumours of the testis had been summarized as follows: [21]
Majority of adrenal rest tumours of testis tend to be detected based upon ultrasound scan screening of patients who have Congenital adrenal hyperplasia. [21]
Some patients manifest with symptoms of congenital adrenal hyperplasia as follows:
Severe forms: It has been stated that patients who have critical adrenal insufficiency with salt losing form do present in shock very early in their childhood. [21]
Milder forms: It has been stated that patients who milder forms of adrenal rest tumour of the testis present with varying degrees of virilization and sexual precocity in a young child, adolescent or adult. [21]
It has been iterated that the commonest manifestation of TTAGS is bilateral presence of palpable nodules within both testes with testicular enlargement. [21]
It has been iterated that even though varying degree of testicular atrophy is common in cases of testicular adrenal rest tumours, manifestation with infertility is rare [21] [38]
Diagnosis
With regard to the diagnosis of testicular adrenal rest tumour, it has been stated that the diagnosis of the tumour is suspected based upon clinical features and ultrasound examination of scrotum [7] [21] and the diagnosis id confirmed based upon pathology examination of specimens of the testis tumour that had been obtained from a biopsy / surgical excision of the testicular tumour. [21]
Laboratory tests
Urine tests
Urinalysis, urine microscopy and culture are routine tests that tend to be undertaken in cases of testicular adrenal rest tumours (TARTS) as part of the general assessment of the patients; however, generally the results would be normal, but if there is any evidence of urinary tract infection, it would be treated appropriately based upon the antibiotic sensitivity pattern of the cultured organism to improve upon the health of the patient.
Haematology blood tests.
Full blood count, and INR are routine haematology blood tests that tend to be undertaken in cases of adrenal rest tumours of the testis but generally the results would tend to be normal; however, if there is any abnormality detected, it would be investigated and treated appropriately to improve upon the general health of the patient.
Biochemistry other tests
Serum urea and electrolytes, CRP, liver function tests, bone profile, and blood glucose, are routine biochemistry blood tests that tend to be undertaken in cases of adrenal rest tumours of the testis but generally the results would tend to be normal; however, if there is any abnormality detected, it would be investigated and treated appropriately to improve upon the general health of the patient
Increased blood levels of 11-β-OH steroids in effluent testicular blood are found in cases of adrenal rest tumours of testis. [21]
Increased 11-β-hydroxylase activity is found within tumour tissue of a patient who has adrenal rest tumour of testis [21]
Low α-fetoprotein, LDH and β-HCG are found in patients who have adrenal rest tumours of testis [21]
Radiology description
Radiology imaging studies that tend to be undertaken in cases of adrenal rest tumours of testis include: ultrasound scan, computed tomography scan, and magnetic resonance imaging scans which would tend to demonstrate the following: [21]
The testis tumours tend to be found to be uniformly hypoechoic with well-defined margins
Multifocally and bilaterality of the tumour is the common finding which tends to be reported in about 5% of the patients.
The mean diameter of adrenal rest tumour of testis upon radiology imaging had been stated to be 16 mm and the diameter of the tumour had ranged between 2 mm and 28 mm, [16]
Prognostic factors
With regard to the prognosis of adrenal rest tumours of testis, it has been iterated that the tumours are uniformly benign in biological behaviour and many of the tumours respond to ACTH suppression by glucocorticoid therapy [21]
It has been iterated that no reports of malignant transformation or recurrence after complete excision of adrenal rest tumour of testis had been documented. [21]
Treatment
The ensuing summations had been made regarding the treatment of adrenal rest tumours of testis: [21]
It has been iterated that early adrenal rest tumours are responsive to glucocorticoid therapy, which suppresses ACTH levels. [21]
It has been stated that surgery is undertaken for tumours that are nonresponsive to glucocorticoid therapy, and for large tumours or if differential diagnosis with other tumours exists. [21]
It has been pointed out that testicular sparing surgery is preferred in order to preserve fertility of patients, as the tumour is benign. [21]
Staging / staging classifications
The staging and staging classifications of adrenal rest tumour had been summarized as follows: [21]
Claahsen-van der Grinten et al. [34] had proposed a staging system for TART development and progression, with an intent to guide treatment of patients as follows:
Stage 1: presence of adrenal rest cells within the rete testis, that are not detectable by scrotal ultrasound
In healthy boys, probably regress in utero or in first year of life
Stage 2: adrenal rest cells may proliferate in the presence of increased concentrations of ACTH (and possibly also of Angiotensin II)
The adrenal rest cells might become visible by ultrasound as one or more small hypoechogenic lesions
Age of onset of cell growth may depend upon the cumulative exposure to ACTH (and angiotensin II) concentrations over time and the number of ACTH (and angiotensin II) receptors on the adrenal rest cells
Complete regression of tumours with ACTH suppression by glucocorticoid therapy is possible
Stage 3: further growth of the adrenal rest cells will compress the rete testis
In pubertal or post-pubertal CAH patients, oligo or azoospermia may already be found due to obstruction of the seminiferous tubules
Signs of gonadal dysfunction: decreased inhibin B and increased follicle stimulating hormone (FSH) and LH levels may also be present
Tumours are responsive to ACTH suppression; nevertheless, the tumours relapse following discontinuation of ACTH suppression
Hence, glucocorticoid therapy is only a temporary solution
Stage 4: further hypertrophy and hyperplasia of the adrenal rest cells with progressive obstruction of the rete testis, leads to induction of fibrosis within the tumour and focal lymphocytic infiltration.
Several small tumours within the rete testis will conflate, forming a single lobulated tumour, separated from the residual testicular tissue by fibrous strands
Differentiation of adrenal cells with loss of ACTH and angiotensin II receptors
Loss of response to ACTH suppression by glucocorticoids
Surgical excision of TTAGS could prevent further decline of testicular function
Stage 5: chronic obstruction subsequently will lead to destruction of the surrounding testicular parenchyma with irreversible damage of the testis
Only indication for surgery is the relief of pain and discomfort caused by TTAGS
It has been pointed out that the aforementioned staging system had not been validated. [21]
It has been iterated that early treatment of CAH with adequate suppression of ACTH levels by glucocorticoid treatment from childhood, might prevent the development of TTAGS. [21]
However, it had also been stated that the benefit needs to be balanced out against the effect of growth suppression by excess glucocorticoid therapy. [21]
Gross description
It has been documented that usually adrenal rest tumour of testis is bilateral, and commonly located in the hilar region of testis. [21]
It has been documented that adrenal rest tumours of testis are well circumscribed yellow to tan nodules that are separated by dense fibrous bands. [21]
Microscopic (histologic) description
The microscopy histopathology examination features of adrenal rest tumours of the testis had been summarized as follows: [21]
The microscopy examination features of adrenal rest tumour of testis, simulates the features of adrenocortical cells. [21]
Microscopy examination of adrenal rest tumour of testis demonstrates: Sheets, cords or lobules of large, polygonal cells with abundant, finely granular eosinophilic cytoplasm, and often with lipochrome pigment. [21]
Microscopy examination of adrenal rest tumour of testis demonstrates: round, giant and hyperchromatic nuclei; nuclear pleomorphism can be prominent with presence of varying sized intranuclear inclusions. [21]
Microscopy examination of adrenal rest tumour of testis demonstrates: thin fibrovascular strands run through the tumour tissue but zonation tends to be absent
Microscopy examination of adrenal rest tumour of testis demonstrates that mitoses are usually absent [21]
Microscopy examination of adrenal rest tumour of testis demonstrates that Reinke crystals are absent and this helps to differentiate adrenal rest tumour of testis from Leydig cell tumours to an extent. [21]
Microscopy examination of adrenal rest tumour of testis tends to demonstrate that the adjacent testicular tissue may be normal or may show evidence of atrophy in the form of reduced / absent spermatogenesis with reduced diameter of the seminiferous tubules, thickened basal membranes and deposition of fibro-hyaline tissue. [21]
The forementioned changes tend to be more marked with tumours of long duration. [21]
Cytology description
The cytology examination features of adrenal rest tumour of testis had been summarized as follows: [21]
Cytology examination of adrenal rest tumour of testis demonstrates: Cellular smears with large polygonal to round cells with foamy cytoplasm, round centrally placed nuclei with prominent nucleoli. [21]
Cytology examination of adrenal rest tumour of testis demonstrates: Abundant lipochrome pigment which is characteristic of adrenal rest tumour of testis. [21]
Cytology examination of adrenal rest tumour of testis demonstrates that Reinke crystals are absent. [21]
It has been pointed out that there is no cytological feature to reliably distinguish adrenal rest tumour of testis from Leydig cell tumour of testis in the absence of Reinke crystals. [21]
Immunohistochemistry & special stains
The immunohistochemistry staining features of adrenal rest tumour of the testis had been summated as follows: [21]
Positive staining:
Positive staining: It has been documented that immunohistochemistry staining studies of adrenal rest tumour testis does demonstrate that the tumour cells exhibit positive staining for CD56 which is strong, and synaptophysin which is strong but can be focal or diffuse [21]
Inhibin: It has been pointed out that immunohistochemistry staining studies for adrenal rest tumours do demonstrate tumour cells that exhibit positive staining for Inhibin but this staining feature is not useful to differentiate adrenal rest tumour from Leydig cell tumour. [21] [39]
Negative staining:
Negative staining: Androgen Receptor: It has been stated that the tumour cells of adrenal rest tumours of testis exhibit negative staining for androgen receptor. [21]
Electron microscopy description
It has been documented that electron microscopy examination studies of adrenal rest tumours of testis do demonstrate abundant smooth endoplasmic reticulum, a moderate number of mitochondria with tubulovesicular cristae, lipid droplets and lipofuscin granules in the polygonal cells. [21]
It has been documented that electron microscopy examination studies of adrenal rest tumours of testis do demonstrate no evidence of Reinke crystals. [21]
Differential diagnosis [21]
It has been documented that Leydig cell tumour of testis is a differential diagnosis of adrenal test tumour and Leydig cell tumour testis has the ensuing features: [21]
Leydig cell tumour of testis: [21]
It has been reported that Leydig cell tumour of testis is less commonly bilateral which does occur in 3% the tumours compared to 75% in TTAGS, [23] [40].
Gynecomastia is more common and this occurs in about 30% of cases, while it is rare in CAH. [23]
Leydig cell tumour is testis is associated with Peutz-Jeghers syndrome, which is not associated with adrenal hyperplasia. [21]
Leydig cell tumour of testis is non-responsive to corticosteroid therapy [21]
Presence of Reinke crystals is diagnostic but had been seen in just 40 of cases. [21]
Malignancy related changes are seen in 10% of Leydig cell tumours of testis while they never occur in TTAGS [21]
In Leydig cell tumour of testis there is absence of adipocyte metaplasia and extensive fibrosis [21]
In Leydig cell tumour of testis, the tumour cells do exhibit Androgen receptor positivity and weak reactivity / negative staining for CD56 and synaptophysin upon immunohistochemistry staining studies. [21]
The biochemical features are of Leydig cell tumour of testis NOT useful to differentiate the tumour from adrenal rest tumour of testis. [21] [23]
[B} miscellaneous narrations and discussions from some case reports, case series and some studies related to neuroendocrine tumours of the testis.
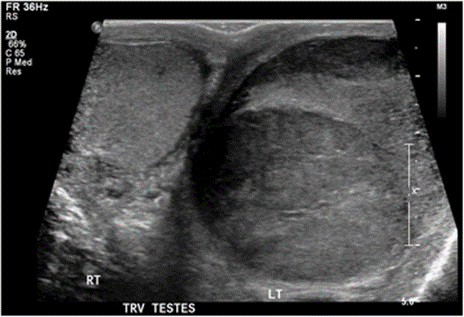
. Heijkoop et al. [41] reported a rare case of neuroendocrine tumour that was metastatic to the testicle, manifesting with testicular mass as an isolated symptom and they described the investigations and management leading them to this uncommon histological diagnosis and they also explored its significance and impact upon further management. Heijkoop et al. [41] made the ensuing educative summations:
Neuroendocrine neoplasms (NEN) include both neuroendocrine tumours (NETs) as well as neuroendocrine carcinoma (NEC).
Distinction between the two-types of neuroendocrine tumours is based upon the level of differentiation and is significant because the tumour behaviour and therefore treatment options differ for the two types of neuro-endocrine tumours.
Well-differentiated and moderately differentiated tumours (WHO grades 1 and 2) are termed NETs with surgical excision the mainstay of treatment, while NECs (WHO grade 3) are poorly differentiated tumours which predict the development of disseminated disease even if localised at the initial time of diagnosis of the tumour and as such are more likely to require adjuvant treatment. [42] [43]
NETs account for less than 1% of all testicular tumours, with primary testicular NETs being more common than a metastasis to the testis. [44]. [45] Nevertheless, neuroendocrine tissue is distributed widely within the body and primary NETs elsewhere within the body have the potential to metastasise irrespective of their size, including many years after the initial diagnosis and treatment of the primary tumour. [6] [46]
Common metastatic sites for the development of NETs include the following: lymph nodes, liver and bone. [46]
The preferred treatment of NETs is surgical excision in view of the fact that they are relatively chemotherapy and radiation resistant; nevertheless, somatostatin analogues and interferon alpha are also utilized in metastatic disease and for the control of symptom where the tumour has functional neuroendocrine activity producing the carcinoid syndrome of flushing, diarrhoea, sweating and bronchospasm. [46] [47]
They were reporting a rare case of metastatic NET presenting with testicular mass as the first symptom and highlight the importance of comprehensive assessment to identify the primary source of this pathology. [41] Heijkoop et al. [41] reported a 59-year-old man who had manifested with a 1-week history of groin discomfort and a palpable left testicular mass. He did not report any systemic features of disease and notably he did not report any symptoms that were consistent with carcinoid syndrome. He had ultrasound scan of his scrotum and testes which demonstrated a 16 mm ×13 mm ×9 mm irregular lobulated hypoechoic mass with internal vascularity within the lower pole of his left testis. The results of his testicular tumour markers were unremarkable. He was planned to undergo a left inguinal orchiectomy, with the undertaking staging CT scan of his chest, abdomen and pelvis prior. He underwent pre-operative CT scan which reported no obvious metastatic disease related to his testicle but the incidental finding of a 3 cm × 2.5 cm ×2 cm soft tissue density mass adjacent his ileo-caecal junction with associated mesenteric tethering. He was referred to the general surgical team for further investigation. He underwent colonoscopy which identified a moderate stenosis of his ileo-caecal valve that was not able to be traversed. Biopsies were taken of this area, histology of examination of the biopsy specimens revealed features based upon which a diagnosis of a well-differentiated, WHO grade 1 NET was made. As was planned, the patient also underwent inguinal orchidectomy with histology of the testicular specimen being multiple separate NETs staining positive for synaptophysin and chromogranin. The testicular NETs measured 3 mm, 10 mm and 40 mm and they were WHO grade 2, originating within the para-testicular soft tissues and extending into the testicular parenchyma. The spermatic cord margin was positive. The Chromogranin A level was 226.Discussion at multidisciplinary team meeting recommended referral to an upper gastrointestinal oncologist and DOTATATE positron emission tomography (PET). DOTATATE PET showed intense tracer uptake at the ileocaecal valve but no avid intra-abdominal lymphadenopathy. He subsequently underwent right hemicolectomy for management of his ileo-caecal tumour, with re-excision of the spermatic cord margin performed concurrently. Intraoperative frozen section pathology examination was used to confirm a clear margin of the re-excised spermatic cord. Final histology examination of the re-excised cord showed chronic inflammation and stitch granuloma only with no residual tumour identified. The ileo-caecal -tumour was confirmed as a grade 2 pT4 N1 (6/18 nodes positive) M1 (previously excised testis) NET of ileal origin. The patient was planned to undergo continued follow-up by the colorectal surgery and upper gastrointestinal oncology teams’ surveillance with six monthly chromogranin A level and DOTATATE-PET scan. The investigations that were undertaken over the course of his follow-up assessment and his management included blood tests (chromogranin A level and testicular tumour markers: alpha-foetoprotein, human chorionic gonadotropin, lactate dehydrogenase), imaging with ultrasound, CT and DOTATATE PET and diagnostic colonoscopy and biopsy. Heijkoop et al. [41] stated that the differential diagnosis of their reported tumour include: Testicular malignancy, neuroendocrine malignancy, synchronous testicular and neuroendocrine malignancies, lymphoma and para-testicular tumours and the treatments undertaken as well as follow-ups planned for the patient include:
Left inguinal orchiectomy.
Right hemicolectomy.
NET with metastasis to the testicle.
Ongoing surveillance with six monthly chromogranin A level and DOTATATE PET scan by colorectal surgery and gastrointestinal oncology teams. Heijkoop et al. [41] made the ensuing discussions:
Metastasis of an NET to the testis was first reported by Cope in 1930, but does remain a rare diagnosis with a recent systematic review identifying only nine prior reported cases. [44]. [48]
They had reported a unique case of isolated metastatic NET manifesting with a testicular mass in an otherwise asymptomatic patient, which they had added to the body of literature on this rare entity and they had raised many important discussion points.
First of all, their reported case is an important reminder of the importance of a thorough workup of any patient who manifests with a testicular mass; as while testicular tumours are a common problem, there are instances where they represent an uncommon pathology.
It has been iterated that in the scenario where the rare diagnosis of testicular NET is made, it is important to distinguish between primary and metastatic NETs of the testis; as while NET metastatic to the testis is rare, the consequences of missed diagnosis of a primary tumour in this setting are significant, with the prognosis of secondary NET in the testis, being far worse than that of a primary testicular NET. [47]
In their reported case, the identification of an ileo-caecal mass upon standard preoperative staging imaging and consequent almost simultaneous histological diagnoses of ileo-caecal and testicular NET made the relationship between the two tumours obvious; nevertheless, in scenarios where staging is undertaken postoperatively or primary tumour is not obviously visible on radiology-imaging, this may be a more challenging conclusion to draw. Consequently, they would encourage the undertaking of gastroscopy and colonoscopy to complete the staging workup in patients who are found to have a testicular NET, especially in the situation where staging imaging appears normal.
Considering the preoperative staging of this case, it is interesting to note that despite the high sensitivity and specificity of DOTATATE PET for low-grade NET, in their patient the avidity which was seen upon DOTATE PET had not identified the lymph nodes metastases separately from the primary tumour. [49] This might be explained by the proximity of the involved lymph nodes to a moderately large primary tumour on preoperative imaging, but raises the question of what the optimal preoperative staging imaging modality for NEN is.
Standard cross-sectional radiology imaging with CT scan is frequently complemented by both MRI scan and nuclear medicine imaging. With regard to nuclear medicine imaging, while previously octreotide Single Photon Emission Computed Tomography (SPECT) scans had been widely utilized, DOTATATE PET had since been demonstrated to be superior to octreotide SPECT in NET, with reported specificity between 88% and 93% and sensitivity of 88%–95%. Nevertheless, it has been stated that in a patient with high-grade or poorly differentiated NEN, that is, an NEC rather than NET, fluorodeoxyglucose-PET is preferable as DOTATATE-PET relies on tumour expression of somatostatin receptors, which reduces with an increased proliferation rate.[50]
They would also remind the reader not to neglect consideration of the testis and other distant sites as potential metastatic sites in patients who are diagnosed as having NET. While less common than metastases to other sites, the testes and other distant sites might not be reliably captured by routine staging and follow-up imaging studies such as CT scan of the chest, abdomen and pelvis, and so may be missed unless the patient undergoes specifically targeted investigations for metastatic disease such as nuclear medicine imaging. In addition, the absence of systemic symptoms suggestive of ‘carcinoid symptom’ should be used cautiously, being a far less useful diagnostic feature than their presence as the carcinoid syndrome is only seen in 20%–30% of patients with metastatic disease. [50]
Finally, taking into consideration the fact that NETs are known to have the potential metastasise many years after initial diagnosis and curative intent treatment, they would recommend that patients who have testicular NET should continue to undergo ongoing follow-up radiology imaging and examination even after they had undergone successful treatment, regardless of whether the testicular NET was primary or metastatic.
The existing literature lacks consensus related to the exact follow-up recommendations but supports both long-term follow-up and utilization of cross-sectional radiology imaging, biomarkers and nuclear medicine imaging in addition to patient history taking and examination. [49] Based upon lessons learnt from their case reports and review of the literature, Heijkoop et al. [41] summated the learning points of the case as follows:
It is crucial for clinicians to differentiate between primary testicular neuroendocrine tumour (NET) and metastasis of NET to the testis, as both are possible, and consequences of a missed primary tumour in a patient who has metastatic NET are significant.
Clinicians should strongly consider utilization of gastroscopy and colonoscopy to complete a staging work up in any patient who has NETs of his testis.
Patients who have NET of testis will require on going follow-up assessments for many years even following their successful treatment in view of the fact that NET has the potential to metastasize even after their initial treatment of curative intent. Reyes et al. [51] studied 10 cases of primary pure testicular neuroendocrine carcinoma. The ages of their 10 patients had ranged between 16 years and 48 years and they had presented with testicular swelling associated with pain or painless testicular mass as well as no history of neuroendocrine carcinoma or any other malignant tumour. All of the patients had undergone orchidectomy. The tumours were low (9 cases) and intermediate (1 case) grades a variegated histopathology appearance which was characterized by a nesting pattern, cords of neoplastic cells with rosettes, or sheets of neoplastic cells. Mitotic activity was absent in 9 cases. In 1 case, mitotic-figures, had ranged from 7 to 8 per high power fields, and cellular atypia and comedo-like necrosis was found present in the tumour. Immunohistochemistry staining studies utilizing a keratin cocktail, chromogranin, synaptophysin, epidermal growth factor, p53, placenta-like alkaline phosphatase, and CD117, (C-kit) were undertaken in all cases. Keratin, chromogranin, and synaptophysin, were positive in all 10 tumours. Clinical follow-up information was obtained for 6 patients with follow-ups which had ranged from 12 months to 60 months and out of these patients, 5 patients had low-grade tumours and they were alive 24 months to 60 months pursuant to their initial diagnosis. One (1) patient who had an intermediate-grade tumour died of his tumour 12 months pursuant to his initial diagnosis. The biological behaviour of these tumours, while in the testicular region, correlated well with the histopathology grade of the tumours. Reyes et al. [51] proposed the replacement of the terminology testicular carcinoid with neuro-endocrine carcinoma, which does better reflect the nature of these neoplasms. Han et al. [52] explores the clinicopathological characteristics and differential diagnosis of primary neuroendocrine tumour (G1) of the testis. Han et al. [52] analysed the clinical, histomorphology and immunohistochemical findings, treatment and prognosis of a patient who had primary neuroendocrine tumour of the testis, and they discussed the relevant literature. Han et al. [52] reported a 52-year-old man who had presented with a painless testicular swelling over the preceding 6 months. He underwent orchidectomy which completely removed the tumour, and histopathological examination of the tumour showed that the tumour cells were arranged in island and beam patterns. The tumour cells were uniform, polygonal and had moderately eosinophilic cytoplasm and fine granular nuclear chromatin. Immunohistochemical staining of the tumour showed that the tumour cells had exhibited positive staining for cytokeratin, CD56, synaptophysin and chromogranin A, and negative staining for inhibin, placental alkaline phosphatase and alpha-fetoprotein. Han et al. [52] stated the following:
Primary neuroendocrine tumour of the testis is a rare tumour which is associated with characteristic radiology imaging features.
The accurate diagnosis of primary neuro-endocrine tumour of the testis depends upon the morphological and immunohistochemical findings.
These tumours should be distinguished from metastatic neuroendocrine carcinomas, teratomas with carcinoid, seminomas, Sertoli cell tumours and granulosa cell tumours.
The treatment of most primary neuroendocrine tumours entails surgical resection combined with other therapies and usually results in a good prognosis.
Wang et al. [53] stated the following:
Testicular carcinoid tumours are rare with only limited studies.
They had identified 29 primary testicular carcinoid cases from 7 academic institutions.
They patients had ranged in age from 12 years to 65 years old and their mean age was 36 years.
The commonest presenting symptom was the sole finding of either a testicular mass or swelling seen in 15 out of 24 cases with available information.
The next most common mode of presentation was as an incidental finding was seen in 6 cases. Two patients had carcinoid syndrome including diarrhoea, hot flashes, and palpitations.
Nineteen of the tumours were pure carcinoid tumours, 3 were associated with cystic teratoma, 2 with cysts lacking epithelial lining, 4 with epidermoid cyst, and 1 with dermoid cyst.
The mean size of the tumour was 2.5 cm.
All 29 primary carcinoids lacked associated intratubular germ cell neoplasia, unclassified type. Mitotic figures were rare within primary carcinoid tumours with only 3 cases showing more than 2 per 10 HPF; necrosis was found in only 1 case. Random scattered mild to moderate nuclear atypia was seen in 12/29 cases. Out of the 28 cases found premortem, their treatment had included focal excision in 3 patients and radical orchiectomy in 25 patients. Follow-up, was available in 24 cases, which had ranged from 1 month to 228 months and their mean follow-up was 52.7 months; of the 20 patients who had testicular typical carcinoid tumours that were found premortem, all were alive at last their follow-up without recurrences or metastases. Out of the 4 patients who had a primary atypical carcinoid tumour, 1 patient at the time of diagnosis had retroperitoneal and lung metastases who after chemotherapy and who underwent resection of the retroperitoneal tumour showing metastatic yolk sac tumour and embryonal carcinoma. After resection, the serum AFP levels had remained elevated and the patient was scheduled to undergo salvage chemotherapy and bone marrow transplant. The other 2 patients who had atypical carcinoid and follow-up had no evidence of disease at 68 months and 114 months. Wang et al. [53] made the following conclusions:
Most primary carcinoid tumours of the testis had a benign clinical course even if associated with epidermoid/dermoid cysts, or histologically mature teratoma.
Nevertheless, lesions that have the morphology of atypical carcinoid can occasionally exhibit metastatic spread. Mweempwa, et al. [54] stated the ensuing:
Merkel cell carcinoma (MCC) is a rare and aggressive neuroendocrine malignancy which often arises in sun-exposed skin and predominantly affects the elderly.
The documented mean ages of MCC at initial diagnosis are 76 years for women and 74 years for men [55]
Immunosuppression caused by solid organ transplant, human immunodeficiency virus (HIV) infection, and B-cell lymphoproliferative malignancies is associated with an increased risk of MCC. [56] [57] [58]
Even though originally thought to arise from Merkel cells in the skin, it has been suggested that MCC might instead originate from skin stem cells or pro-/pre- and pre-B cells. [59] [60]
Merkel cell polyoma virus (MCPyV), which is a double-stranded deoxyribonucleic acid (DNA) virus, had been linked to the development of MCC [61] and may have a role to play in the transformation of pro-/pre- and pre-B cells into MCC.
MCC more commonly occurs in the skin of the head and neck, followed by the upper limbs and shoulders.
It is rare for MCC to have an unknown primary site. [55]
MCC metastasizing to the testis is rare.
Up to the time of the report of their article, there were seven cases of testicular MCC published in the literature. [62] [63] [64] [65] [66] [67] Mweempwa, et al. [54] reported a 66-year-old Maori man who had presented to their hospital with a history of an enlarged left testicle of 10 weeks’ duration. His clinical examination demonstrated a non-tender swelling of his left testicle. His alpha-fetoprotein (AFP) and beta-human chorionic gonadotropin (BHCG) levels were within normal limits. His lactate dehydrogenase (LDH) concentration was mildly raised at 267 U/L (normal range 120–250 U/L). He had ultrasound scan of his testes which demonstrated an enlarged left testicle that measured 7 cm × 5.5 cm × 4.3 cm with an estimated volume of 87 ml. A large, heterogeneous mass was demonstrated which had involved the entire testicle with increased vascularity (see figure 1). The patient’s right testicle measured 3.7 cm × 2.5 cm × 1.8 cm with an estimated volume of 8.8 ml. He underwent left orchidectomy. The macroscopic specimen consisted of well-circumscribed nodular lesions of various sizes, with the largest measuring 45 mm × 15 mm and had contained solid and gelatinous components (see figure 2). Sections of the specimen showed tumour that was composed of sheets of small, blue, round cells divided into nodules by fibrous septae (see figure 3). Immunostaining of the tumour showed that the tumour to be cytokeratin 20 (CK20)-positive with a typical paranuclear dotlike staining pattern (see figure 4). The stain also showed that the tumour cells had exhibited positive staining result for CD56, a neuroendocrine marker (see figure 5), as were CD117 and CK (paranuclear dots). The immunohistochemistry staining result was negative for CK7, placental alkaline phosphatase (PLAP), CD30, CD20, AFP, S100, SOX10, prostate-specific antigen (PSA), chromogranin, and thyroid transcription factor 1 (TTF-1). The Ki-67 level was >50 %. This immunostaining pattern raised the possibility of metastatic MCC.
Macroscopic image of the left testis. The left testis consists of well-circumscribed nodular lesions of varying sizes containing solid and gelatinous components. Scale bar = 20 mm
High-power image (hematoxylin and eosin-stain, original magnification ×400) showing small, blue, round cells. Tumor nuclei have a neuroendocrine appearance with speckled chromatin. A residual seminiferous tubule is seen at top right Positive result for cytokeratin 20 with a typical dotlike pattern of staining (original magnification ×200)
Positive result for CD56, a neuroendocrine marker (original magnification ×200)
The patient again remanifested in their hospital 3 months later this time with a right testicular mass. The results of his tumour markers, including LDH, AFP, and BHCG, were within normal limits. He had ultrasound scan radiology imaging which demonstrated a new lesion within his right testis that measured 3 cm × 2.6 cm × 2.3 cm. He underwent a right orchidectomy, and sections of the specimen showed diffuse infiltration of small, blue, round cells. Immunostainings of the tumour were positive for CD117, CD56, synaptophysin, CK20 (dotlike), and cytokeratin AE1/AE3 (dotlike). The tumour cells were negative for inhibin, PLAP, PSA, S100, CD30, CD45, CD3, CD20, TTF-1, and napsin A. The Ki-67 level was 80 %. This pattern was adjudged to be consistent with a poorly differentiated neuroendocrine tumor in keeping with metastatic MCC. A primary site of the tumour was not identified, and a staging computed tomographic scan did not demonstrate evidence of other metastases. The patient did receive six cycles of adjuvant carboplatin and etoposide chemotherapy. He remained disease-free 18 months pursuant to his completion of chemotherapy.
Mweempwa, et al. [54] made the ensuing summating discussions
It had been suggested that the testes are a sanctuary site for Merkel cell tumours.
It is conjectured that the occurrence of MCC metastases in the testes soon after completion of adjuvant chemotherapy could be due to the presence of a blood-testis barrier that prevents the eradication of tumour cells with chemotherapy agents, thus making the testes a sanctuary site [66]
There are also reports of isolated testicular recurrences in haematological malignancies, which had suggested that the testes are a sanctuary site. [68] [69]
Nevertheless, in a retrospective analysis of patients who had metastatic germ cell tumours who were treated with primary chemotherapy, 43 % had no viable tumour within the testes following delayed orchidectomy, which showed that adequate concentrations of chemotherapeutic agents could be achieved despite the presence of a blood-testis barrier. [70]
It is interesting to note that their reported patient had developed testicular MCC in both testes, with an unknown primary site. It is possible that the primary tumour was eradicated through immunological surveillance, whereas the blood-testis barrier allowed tumour cells in the testes to escape this and to proliferate. Another possibility is that the primary and metastatic disease were heterogeneous and had different properties.
Immunohistochemistry study analysis does help to differentiate testicular MCC from other malignancies such as metastatic small cell carcinoma of the lung, small cell phenotype melanoma, and lymphoma. Paranuclear dot positivity for CK20 and CAM5.2 is a typical feature of MCC. [71] MCC is also positive for CK AE1/AE3 and neuroendocrine markers, synaptophysin, and chromogranin. Positive CK20 and negative TTF-1 and CK7 distinguish the tumour cells from small cell lung carcinoma [72]. Positive staining for S100, HMB-45, and Melan-A is characteristic of melanoma. Negative leukocyte common antigen (CD45) differentiates MCC from lymphoma.
Treatment of early stage MCC is with the undertaking of surgical resection with 1 cm to 2-cmtumour free margins and sentinel lymph node mapping and biopsy if there is no clinical evidence of regional lymph node involvement. Radical lymphadenectomy is recommended if the regional lymph nodes are involved [73]. Alternatively, definitive lymph node irradiation can be undertaken because this had been demonstrated to have efficacy similar to completion lymphadenectomy, with no difference in overall survival [74].
In a retrospective analysis of 6908 patients with MCC, adjuvant radiotherapy to the resection site was associated with improved overall survival for stage I-II MCC, whereas adjuvant chemotherapy did not improve overall survival for stage III MCC [75].
In another population study of 4815 patients who had head and neck MCC, adjuvant chemoradiation and radiation therapy had conferred better overall survival than surgery alone. A survival benefit was also demonstrated in the adjuvant chemoradiation group compared with the adjuvant radiotherapy group in patients who had tumour sizes of at least 3 cm, positive margins, and male sex [76].
Chemotherapy regimens for metastatic MCC are often similar to the ones that are utilized for high-grade neuroendocrine cancers and small cell cancers, with a number of agents showing activity, including combinations of cisplatin, carboplatin, etoposide, cyclophosphamide, doxorubicin, vincristine, bleomycin, and 5-fluorouracil. MCC is regarded as chemotherapy-sensitive, but the duration of response is short.
In one series, the response rates to chemotherapy were 57 % for metastatic MCC and 69 % for locally advanced MCC, with median overall survival of 9 months and 24 months, respectively [77].
The main predictor of survival in MCC is tumour stage at the time of diagnosis. Male sex and tumour size greater than 2 cm are unfavourable factors. [55]. It has been suggested that low tumour depth, absence of lympho-vascular invasion, a nodular growth pattern, and intra-tumoral lymphocyte infiltration are associated with a lower risk of death [78] [79]. It has also been noted that patients who have MCPyV DNA-positive tumours have a favourable prognosis compared with those with MCPyV DNA-negative tumours [80].Mweempwa, et al. [54] made the ensuing conclusions:
They had reported a rare case of a patient who had testicular MCC occurring twice with an occult primary tumour.
Even though a treatment guideline for MCC had been proposed [73], there is no guidance on how to manage testicular MCC.
Current treatment is individualized and depends upon the extent of disease at the time of the initial diagnosis.
Orchidectomy is the initial management modality if the disease is localized to the testes, but the role of adjuvant therapy is yet to be determined.Abbosh et al. [81] stated the following:
Carcinoids are neuroendocrine tumours and which most frequently occur within tissues that are derived from the embryonic gut.
These carcinoid tumours can occur in any organ site but are rare in the testis. The cell type giving rise to testicular carcinoid is unknown.
They postulated that testicular carcinoid may have a germ cell origin.Abbosh et al. [81] reported their analysis of protein and genetic markers of germ cell neoplasia, using immunohistochemistry and fluorescence in situ hybridization, in four testicular carcinoid tumours. Abbosh et al. [81] reported the results as follows:
All four cases of testicular carcinoid tumour had arisen within a background of mature teratoma.
Isochromosome 12p was identified in carcinoid tumour cells in all four samples. 12p overrepresentation was also observed in three cases. Isochromosome 12p and 12p overrepresentation were present in cells of coexisting mature teratoma in three cases. Carcinoid tumours showed strong immunoreactivity for synaptophysin and chromogranin, but no immunoreactivity for OCT4, CD30, c-kit, TTF-1, and CDX2. Membranous and cytoplasmic staining for β-catenin was detected in three cases.Abbosh et al. [81] concluded that their findings suggest that testicular carcinoid represents a phenotypic expression of testicular teratoma and is of germ cell origin.Son et al. [82] stated the following;
Carcinoid tumours are derived from neuroendocrine cells and they could release serotonin as well as other vasoactive substances into the systemic circulation emanating in the development of carcinoid tumour.
Carcinoid tumour of the testis is an uncommon disease which does account for less than 1% tumours of the testis, and carcinoid tumour of the testis rarely manifests symptoms of carcinoid syndrome. Son et al. [82] reported a case of carcinoid syndrome which had arisen from a primary carcinoid tumour of testis. Son et al. [82] reported a 21-year-old gentleman, who had manifested with facial flushing and diarrhoea for 5 years. He had an enlarged left testis as well as a 1 cm, ill-defined, hard, non-tender mass within his right testis. His 24-hour urinary excretion of 5-hydroxyindoleacetic acid level was raised at 16.1 mg/day. He had a somatostatin scintigraphy which correlated with carcinoid tumour within both his left and right testis. Pursuant to his undergoing of bilateral orchidectomy, the patient’s facial flushing as well as his diarrhoea had stopped.
Neely et al. [83] made the following iterations:
Testicular carcinoid tumours are very uncommon and they rare and account for less than 1% of all testicular neoplasms. [84]
These tumours may be classified into three distinct groups, most commonly (1) primary testicular carcinoid, (2) carcinoid differentiation within a mature teratoma, and (3) metastases from an extra-testicular source.
Carcinoid tumours of the testis do not follow the age category of men who are affected most commonly by germ cell tumours, whose ages range had ranged from 20 years to 40 years, and also these types of cases had been reported in males with their ages ranging from ten years to eighty-three years. [85]
Manifestation of carcinoid tumours may be with self-detected testicular mass or testicular ache as with common testicular tumours, or uncommonly with carcinoid syndrome
They were reporting a case of primary carcinoid tumour of the testis without features of carcinoid syndrome.Neely et al. [83] reported a 29-year-old male who manifested in their outpatient clinic with a several weeks-history of a painless right sided testicular swelling which he found on his self-examination. He did not have any recent history of trauma, urinary tract or sexually transmitted infections. Upon his examination he was found to have a hard mass within the upper aspect of his right testis which felt strongly suggestive of testicular tumour. He had an ultrasound scan of his scrotal contents which showed normal appearance of the left testis and a mixed echogenic mass which had occupied the majority of the superior aspect of his right testis. The mass measured 3.5 cm × 2.3 cm and had increased vascularity (see figure 6). Tumour markers (beta human chorionic gonadotrophin, alfa-fetoprotein and lactate dehydrogenase) were normal and a pre-operative chest radiograph did not reveal any metastases. The patient underwent radical orchidectomy and insertion of testicular prosthesis soon after diagnosis.
Figure 6: Ultrasound image of the right testis showing mixed echogenic mass in upper pole in keeping with testicular tumour Reproduced from: [83] Histological analysis of the tumour showed a well circumscribed tumour composed of monomorphic cells arranged in a nested trabecular pattern. The tumour cells had granular chromatin and scarce mitotic figures. Immunohistochemistry of the tumour showed that the tumour cells had exhibited strong positive staining with chromogranin (see figure 7), synaptophysin (see figure 8), CD56 and PGP9.5. These features were adjudged to be in keeping with a well differentiated carcinoid tumour with no teratoma components or other germ cell elements. There was no lympho-vascular invasion, the tumour was noted to be confined to the testis and had a low proliferation index.
Figure 7: Monomorphic cells in nested trabecular pattern. Staining strongly positive with chromogranin Reproduced from: [83]
Figure 8. Cells staining strongly positive with synaptophysin Reproduced from [83]
The patient subsequently underwent a staging computed tomogram (CT) of chest, abdomen and pelvis, which demonstrated no significant para-aortic or iliac lymphadenopathy and no pulmonary abnormality. Multiple mesenteric nodes were identified which were not said to be typical of spread of testicular tumour.
The patient was referred to a specialist gastroenterologist for further investigation to exclude the possibility of testicular metastasis from an extra-testicular primary carcinoid. He had NM octreotide scan with SPECT which showed two to three subtle foci of activity towards the dome of the right lobe of his liver in segment four and eight. These were adjudged not to be definite for liver metastases; nevertheless, these could not be excluded. No lesions were found to be over expressing somatostatin receptors. His subsequent assessment with MRI had confirmed that the areas of abnormality within the liver were consistent with simple cysts.
Neely et al. [83] made the ensuing discussions:
Testicular carcinoid tumours are a rare entity and they are almost never suspected preoperatively.
The majority of carcinoid tumours of the testis are only diagnosed based upon histopathology as they do not become clinically apparent until there is metastatic spread or the presence of carcinoid syndrome. [85]
Carcinoid tumours do arise from neuroendocrine cells; nevertheless, the presence of neuroendocrine cells had not been reported within the testis, leaving the origin of primary testicular carcinoid debatable. [86]
Several cellular origins of these tumours had been postulated. Mai et al. [87] found that the origin of testicular carcinoid tumours was located within the same progenitor cell from which Leydig cells derive. [87]
Merino et al. [86] supported the possibility of a germ cell origin, finding intra-tubular germ cell neoplasia within the testicular tissue surrounding a pure carcinoid [86]
Hence, primary testicular carcinoid may be the remaining component of a burnt out teratoma or due to a one-sided development of teratoma. [86] 88]
Carcinoid syndrome is a presentation of carcinoid tumours and does occur as a result of the action of vasoactive tumour products.
The syndrome is uncommon, and it occurs in about 10% of patients [88] typically only once the tumour had metastasised to the liver or lungs.
Serotonin is the commonest tumour product and when released into the systemic circulation it causes the symptoms of carcinoid syndrome. [89] These symptoms include increased gastro-intestinal motility, bronchoconstriction, vascular constriction and dilatation. [89]
Serotonin is metabolised to 5-hydroxyindoleacetic acid (5-HIAA) which could be measured in the urine. Any patient who has vasoactive symptoms and a testicular lump should have 24 hour urinary 5-HIAA examination performed prior to his surgery.
It has been stated that considering that almost 10% of testicular carcinoids are metastases from another location, [85] it is essential to thoroughly investigate these patients in order to find or exclude an extra-testicular primary source. A multimodal approach has been recommended. Barium contrast studies and CT scan may identify mucosal thickening or luminal narrowing to suggest bowel involvement. CT scan is also good for the detection of mesenteric extension of the tumour and presence of liver metastases [90]
Somatostatin receptor scintigraphy utilizing indium-111 labelled octreotide is now superior to CT in localisation of primary tumour site and has a sensitivity for the detection of metastases of up to 96%. [90] It has superseded meta-iodobenzylguanidine scanning which has a sensitivity of 50% for detecting metastases. [90] These investigations also do serve to detect synchronous tumours, as it is known that carcinoids have a high rate of second primary malignancy.[89]
The undertaking of radical orchidectomy is curative for testicular confined carcinoid tumours. [88]
The long-term prognosis of carcinoid tumours is dependent upon the size, association with teratoma and presence of metastases.
Zavala-Pompa et al. [91] showed that larger tumours (that measure 7.3 cm versus 2.9cm) and the presence of carcinoid syndrome predicted increased metastatic potential and hence poorer prognosis. [91]
The prognosis of carcinoid tumours that arise within teratoma is better than pure testicular carcinoid. [91]
There had been several reports of carcinoid tumours causing delayed metastases, in one case the metastasis was diagnosed 17 years after the initial diagnosis, which highlighted the need for long-term follow-up. [92]
Patients should undergo biochemical and radiological imaging follow-up; nevertheless, the frequency and duration of follow-up had remained open debate
Sutherland et al. [93] had suggested that patients should undergo three monthly 5-HIAA measurements for the first year after the diagnosis and annually thereafter. [93]
More recent guidelines suggested that urinary 5-HIAA levels do not accurately correlate with disease progression and metastases may occur in the absence of an elevated urinary 5-HIAA.
Serum chromogranin A (a glycoprotein secreted by carcinoid tumours) had been reported to be a sensitive and specific marker which might correlate with relapse in gastrointestinal carcinoids.[90] It may also be of use in the follow-up assessment of testicular carcinoids, a fast-rising level being associated with a poor prognosis.
Neely et al. [83] made the following conclusions:
Testicular carcinoid tumours are very rare.
It is imperative that once a testicular carcinoid tumour has been diagnosed, the patient should undergo thorough investigation to exclude an extra-testicular primary and metastases.
Long-term biochemical and radiological imaging follow-up is essential given the potential for delayed metastases.
Takada et al. [94] stated the following:
Primary testicular carcinoid tumours (TCTs) are very rare, and the tumours tend to large tumours with regard to size, and presence of carcinoid syndrome in association with these tumours does predict a malignant course.
Histologically, it is difficult to differentiate between benign and malignant testicular carcinoid tumours (TCTs).
Takada et al. [94] reported a case of a primary pure TCT with an unusual manifestation in a 21-year-old gentleman, who had an asymptomatic, enlarged testis on the right side of his scrotum for y years. Upon gross examination, the tumour had measured 9.6 cm in diameter. His Ki-67 index labelling index was 19.8%. He underwent orchidectomy and 30 months pursuant to his orchidectomy, the patient had remained asymptomatic.
Lubana et al. [95] stated the following:
Neuroendocrine tumours were first described by Langhans in 1867. [96]
The terminology carcinoid (Karzinoide) was coined by German pathologist Oberndorfer in 1907.
Cope in 1930 had described the first case of metastatic carcinoid tumour which had metastasized from small bowel. [48]
In 1954, Simon et al. reported the first case of primary testicular carcinoid. [97]
Primary testicular tumours are uncommon and they constitute 0.23% of all testicular tumours.
Testicular carcinoid tumours (TCT) have a mean age at presentation of 46 years and the ages at presentation had ranged between 10 years and 83 years).[47]
Even though since 1930 more than 60 cases of testicular carcinoid had been reported, it still remains a very rare diagnosis.
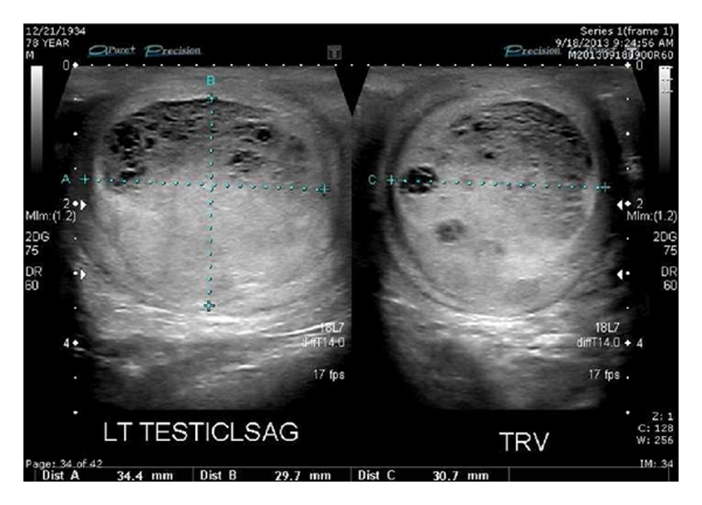
In their report, they had described a primary testicular carcinoid tumour of the testis and they had reported an extensive literature review to cover all the aspects of carcinoid tumour including the definition, classification, origin, presentation, diagnostic evaluation, management, prognosis, and follow-up. Lubana et al. [95] reported a 34-year-old man with no past medical history, who had presented with right scrotal swelling for one year, with recent onset of pain. He did not have any history of testicular trauma, haematuria, undescended testis, systemic symptoms, or weight loss. He did not have any family history of testicular cancer. His clinical examination revealed an enlarged tender mobile right testicular mass. He had ultrasound scan of his scrotal contents which showed an enlarged right testis, which was heterogeneous in echo texture that measured 5 cm ×4.4 cm × 4.8 cm, with focal testicular parenchymal hypoechoic mass that measured 1.7 cm ×1 cm ×1.6 cm, which was suspicious for neoplastic process (see figure 9).
Figure 9: Ultrasound showed enlarged right testis, heterogenous in echotexture (5.0x4.4x1.6 cm). Reproduced from [95] Under Creative Commons Attribution Agreement License. The results of his serum Beta human chorionic gonadotropin [β-HCG] and alfa-fetoprotein [AFP] were normal with elevated lactate dehydrogenase (LDH) 401 (90–225) U/L. He had staging computerized tomography (CT) scan which did not show any evidence of metastasis or adenopathy. The patient underwent radical orchidectomy. Gross examination of the specimen demonstrated that, the right testicle and epididymis was covered by intensely fibrotic tunica vaginalis. The testicle was entirely occupied by the tumour that measured 4.5 cm ×4.5 cm ×4 cm, with 90% necrosis. The tumour was found to be confined to the testis and epididymis without lympho-vascular invasion.The histology of the tumour was consistent with a well-differentiated neuroendocrine carcinoma. The histology demonstrated nests of monotonous tumour cells with relatively abundant eosinophilic cytoplasm, round to oval nuclei, distinct nuclear membrane with “salt and pepper”-like chromatin (see figure 10). Immunohistochemistry studies of the tumour showed that the tumour cells had exhibited positive staining with chromogranin, synaptophysin (see figure 11), cytokeratin AE1/AE3, and CAM5.2 and negative for placental alkaline phosphatase, CD30, β-HCG, AFP, and epithelial membrane antigen. Ki-67 labelling index was less than 1% of tumour cells. The final diagnosis was carcinoid tumour which was localized within his testis. The cancer was classified as pT1 N0 M0 S2 [LDH 401 U/L] as per the American Joint Committee on Cancer (AJCC) TNM staging for testicular cancers.
Figure 10: (A) Low-power view (40x): Showing nests of tumour cells with surrounding abundant blood vessels. (B): High power view (400x); Monotonous tumour cells with relatively abundant cytoplasm, distinct nuclear membrane with “salt and pepper” -like chromatin. Reproduced from [95] Under Creative Commons Attribution License.
Figure 11: Positive Synaptophysin staining of right testis in primary carcinoid tumour. Reproduced from [95] Under Creative Commons Attribution License. The possibility of an extra-testicular carcinoid tumour was excluded with negative esophagogastroduodenoscopy and colonoscopy. A nuclear octreotide scan which was undertaken showed focal radiotracer activity projecting over the scrotum (benign physiologic variant); nevertheless, an octreotide avid tumour could not be excluded. The rest of the body demonstrated no evidence of octreotide avid tumour. The results of his urinary excretion of 5-hydroxyindoleacetic acid (5-HIAA) and chromogranin A were within normal range.Lubana et al. [95] made the ensuing educative discussions:
Carcinoid tumours are neuroendocrine tumours which arise from enterochromaffin/Kulchitsky cells.
These cells are widely distributed throughout the body.
Nevertheless, the carcinoid tumours are uncommon outside the gastrointestinal tract (65%) and respiratory tract (25%) and they are very rarely found in the testis, which could be primary or metastatic.[98]
The neuroendocrine neoplasms are currently defined into 3 groups by the WHO/ European Neuroendocrine Tumor Society (ENETS). The classification is based upon the immunohistochemistry staining of Ki-67 or mitotic count – Neuroendocrine Tumours G1 (NET) G1: Ki-67 <2>20%. The term “carcinoid” is used for NET G1. [99]
The histogenesis of pure testicular carcinoma had not been well established. It was described that testicular carcinoids typically occur within the background of teratoma (a germ cell neoplasm) giving the rationale that testicular carcinoid tumour could be of germ cell origin, which was further supported by immunohistochemistry and Florescence in situ hybridization (FISH) techniques utilized by Abbosh et al. They found that Isochromosome 12p and 12p overrepresentations were present in both the carcinoid tumour cells as well as in the cells of co-existing mature teratoma.[81]
Teratoma could give rise to TCT by various mechanisms. The testicular carcinoid might be a component of teratoma with regression of rest of the elements. The other mechanism is the preferential development of Argentaffin cells in teratoma. Nevertheless, Argentaffin cells are not found within the testis but germ cells could give rise to any cell type due to their totipotency [100].
The ovarian carcinoid tumour arising as a component [101] or as a malignant transformation [102] of mature cystic teratoma had been published. Based upon this analogy similar mechanism is assumed to occur in primary TCT on literature review. Nevertheless, carcinoid tumour had never been reported to be arising as a component or resulting from the malignant transformation of teratoma [87].
It has been postulated that Leydig cells may be the origin of TCT due to their neuroendocrine features. Mai et al. [87] had demonstrated the presence of transitional cells expressing features of both the Leydig cells and carcinoid tumour cells in primary TCT. Also, one out of nine Leydig cell tumours in their study showed neuroendocrine differentiation. These findings had supported that both carcinoid tumour cells and Leydig cells may have the same progenitor cell origin in the primary TCT [87]
The commonest manifestation of TCT is painless testicular enlargement, followed by testicular pain, hydrocele and very rarely cryptorchidism. The duration of symptoms of TCT could last as long as 240 months. The left testis is most commonly involved with only one report of bilateral involvement of testis [91]. When these tumours are associated with systemic symptoms including episodic wheezing, flushing, diarrhoea, the terminology carcinoid syndrome is used.
Grossly the tumour appears solid with yellow to tan colour with firm texture due to excessive desmoplasia. The calcifications could be seen. The average of the tumour is 4.6 cm and the tumour size had varied between 1.0 cm to 9.5 cm. Microscopically, the tumour cells are composed of monotonous polygonal-shaped cells with eosinophilic cytoplasm and finally dispersed chromatin within the uniform bland nuclei. The neoplastic cells could be found to have different architectural arrangements but trabecular and insular patterns predominate. The necrosis can be seen in large size tumours. Mitotic figures are rarely seen upon microscopy examination of the tumours. [81] [103]
The diagnosis should be provided with clinical examination of the scrotum in a patient with chronic painless swelling. Doppler ultrasound scan is the initial test of choice. The diagnosis is made with the help of tumour biomarkers, CT-Scans, Magnetic Resonance Imaging (MRI), Nuclear medicine techniques like 111 In-Pentetreotide Scintigraphy, 131 MIBG (Meta-Iodobenzylguanidine) and endoscopy.
5-HIAA is a good initial test for the diagnosis and it has a high specificity (100%) but poor sensitivity (<35>
Patients who have TCT very rarely can express features of carcinoid syndrome but only if there is metastasis to liver or lungs [105]. Therefore, any patient who manifests with symptoms of serotonin excess and testicular swelling must have 24hr urinary 5- HIAA prior to undergoing surgery [83]. Due to the fact that it is difficult to suspect testicular carcinoid pre-operatively and 5 HIAA taken prior to surgery will serve as the baseline tumour marker if the tumour turns out to be carcinoid [93]. Platelet serotonin is the sensitive marker for detection of carcinoid tumour especially if the carcinoid tumours have low serotonin production. This makes platelet serotonin a reliable tool for early diagnosis of the carcinoid tumour and also an excellent marker for the identification of residual tumour following surgery [106].
It is important to meticulously investigate for primary since 10% of the TCTs do have extra-testicular primary tumour. The diagnosis of primary TCT is made only after the exclusion of extra-testicular primary since the morphological and histological appearance of the primary and metastatic TCT is the same [91].
Staging CT scan is used for the detection of metastasis. 111 In-Pentetreotide scintigraphy has a sensitivity of 80% to 90% to localize the tumour and could also be used to predict the response to octreotide therapy. 131-MIBG has a lower sensitivity than scintigraphy scan. Sensitivity in detecting the tumour could be increased to 95% by combining both the scans. When bone metastases are suspected bone scintigraphy should be used since it has a higher sensitivity of 90% to 100% than 111 In-Pentetreotide scintigraphy, which has a sensitivity of 50%, and 131 I-MIBG scan which has a sensitivity of 20%. Video capsule endoscopy is a more advanced technique for identifying primary carcinoid tumour in the small bowel and thus early resection [98]. It is a reasonable small bowel imaging technique since carcinoid tumours are mostly found in the ileum [47].
Radical orchiectomy is the treatment of choice. Carcinoid tumours in general have a very poor response to chemotherapy or radiotherapy. Symptomatic treatment should be given to the patients with carcinoid syndrome. Octreotide, a somatostatin analogue inhibits the release of hormones and neurotransmitters and thus causing symptomatic improvement in about 80% of the patients [98]. The slow-release preparations of somatostatin analogues are available (Sandostatin LAR(R) Depot).
The octreotide has variable antiproliferative effect ranging from partial to complete regression of metastatic carcinoid tumour. Leong and Pasieka reported 2 cases of metastatic carcinoid tumours where octreotide was given for symptomatic treatment before the debulking surgery. Previously seen radiographic metastatic lesions were found to be regressed completely during laparotomy [107].
In view of improved prognosis and longer survival time due to advanced diagnostic and therapeutic techniques other late complications involving metastasis to skin and skeletal system and carcinoid heart disease (CHD) had been reported [108] [109].
Long term follow up is recommended due to the fact that delayed metastasis had been reported even up to several years with one case of metastasis occurring after 17 years following his initial diagnosis [100].
Multimodal approach treatment could be tried where tumour recurrence is highly suspected. Since there are no standardized protocols for the follow up, annual clinical examination and frequent Urinary 5HIAA assessment is a reasonable approach. CgA is of particular importance when the 5 HIAA comes out to be normal in a patient after orchiectomy, since normal 5 HIAA does not exclude the presence of tumour recurrence or metastasis [47]. Sutherland et al. purposed 3 monthly follow up with 5 HIAA measurement for the first year and then annually [83]. Use of platelet serotonin is valuable in detecting residual tumour pursuant to resection of tumour [106]. If still there is any doubt of tumour recurrence or metastasis, 111 In-Pentetreotide scintigraphy could be used [47].
Carcinoid tumours generally have portended excellent prognosis due to their indolent course.
Aggressiveness of the carcinoid tumours is directly proportional to the tumour size of higher than 7.3 cm and evidence of carcinoid syndrome [83]. The prognosis of carcinoid tumour depends upon the extent of tumour spread. 5-year survival rate for localized disease is 93% and for distant metastatic disease is 20–30% [98]. Only 3 deaths had been reported related to distant metastasis [91].
Since there is no effective treatment available for metastatic lesion(s) the prognosis is poor. So far there is only one case report where the metastatic lesion was removed by surgical therapy. No new metastases were found after 25months of follow up.[110] Lubana et al. [95] made the following conclusions:
Their reported case had added to the rare reports in the literature of a primary carcinoid tumour of the testis having low malignant potential.
Due to the advanced diagnostic and therapeutic modalities, the survival time has been prolonged but long-term complications like CHD and bone and skin metastasis have emerged.
Serotonin levels could be elevated even in the absence of carcinoid syndrome. The elevated levels of serotonin could cause CHD leading to reduced survival and poor quality of life; therefore, it is important to follow the hormone levels.
High-risk cases with tumour size measuring more than 7.3 cm, presence of carcinoid syndrome, poorly differentiated tumours and tumours with invasion should be followed more closely for recurrence and development of metastasis. This should be done by monitoring biochemical markers on a regular basis and in case of doubt, scintigraphy should be performed. The surgery should be performed if the metastatic lesion(s) is/are resectable and if not accessible for resection, a trial of octreotide therapy could be given due to its antiproliferative and antihormonal properties. It is important to exclude metastasis in case of testicular carcinoid tumour since the morphology and histology cannot distinguish the primary verses metastatic. Blumberg et al. [111] stated that Carcinoid tumour of the testis represents only 0.23% of all testicular malignancies. Blumberg et al. [111] reported the first case of bilateral asynchronous carcinoid tumour of the testis with a primary site in the small bowel. Blumberg et al. [111] reported a 49-year-old man who had presented with several months of painless enlargement of his left testis. He underwent orchidectomy which revealed carcinoid tumour. The patient presented to the emergency room 2 weeks later and he was found to have small bowel obstruction due to an ileal mass. The resected mass was the primary source of the carcinoid tumour. Four years later, his examination revealed carcinoid tumour within his remaining testis.Martínez Ballesteros et al. [112] reported a case of a carcinoid tumour originated in a testis which was associated with mature teratoma in a 31 years old male. Martínez Ballesteros et al. [112] stated the following:
Primary gonadal location of this tumour is unusual, moreover when the tumour is associated with teratoma.
Early diagnosis and treatment determine the prognosis of the patients who are affected by this tumour since the only curative potential treatment is surgery.
Follow up must be extent for years due to the possibility of late relapse. Prunoiu et al. [113] reported a 39-year-old patient who was admitted in their department, for a non-painful abdominal tumour and concomitant testicular tumour. The results of his serum tumour markers (-human chorionic gonadotropin, alpha-fetoprotein and lactate dehydrogenase) were within normal limits. Lung and bone metastases were diagnosed upon his CT scan. The histopathological diagnosis consisted of immunohistochemical study of his orchidectomy specimen as well as of the bioptic material from bone marrow puncture. Prunoiu et al. [113] made the following conclusions:
The diagnosis of testicular carcinoids is based upon immunohistochemistry study.
Radical orchidectomy is the only potentially curative treatment for this type of malignancy.
Adjuvant chemotherapy determined size reduction of the lung and bone metastases and the disappearance of the lymph node metastases. Darré et al. [114] reported a- 29-year-old man, without specific ascendants, who was seen in the urology department for progressive scrotal swelling of 6 months, associated with pain. After surgery, histology examination of testicular tumour showed diffuse tumour proliferation which consisted of small round monotone cells with hyperchromatic nuclei evoking undifferentiated carcinoma. Immunohistochemistry staining of the tumour showed that tumour cells were positive for chromogranin A and negative for placental alkaline phosphatase and α-fetoprotein. Darré et al. [114] concluded that primary neuroendocrine carcinoma of the testis is a very rare malignant tumour. Immunohistochemistry contributes to its diagnosis in relation to other metastatic neuroendocrine carcinomas, carcinoid tumour teratomas, seminoma, and Sertoli cells.
Penke [115] made the ensuing iterations:
Nneuroendocrine tumours (NETs) of the testis are very rare.
Neuro-endocrine tumours (NETs) comprise 0.2% to 1% of the tumours of the testis. [116] [117]
About 60 cases had been reported in the literature. [91] [117]
Their own search of the literature published between 1976 and May 2014 resulted in 51 securely identifiable patients with a primary NET of the testis which were published as casuistry or small patient series as well as other, not clearly identifiable, case presentations.
Out of the 51 identified patients, 37 were aged between 10 years and 83 years with an average age of 48 years (partly calculated as average of a patient series).
The individual age was mentioned in 30 of these patients who were divided up into the life decades as follows: 10 years to 20 years: 2 patients, 20 years to 30 years: 5 patients, 30 years to 40 years: 5 patients, 40 years to 50 years: 5 patients, 50 years to 60 years: 5 patients, 60 years to 70 years: 2 patients, 70 years to 80 years: 5 patients, and >80 years: 1 patient.
The largest published series until the day of publication of the article was with 29 cases [53] that had been reported an average age of 36 years with the ages ranging from 12 years to 65 years of patients with a NET of the testis.
Only a small part that constituted 3% to 13% of the primary NETs of the testis is associated with a carcinoid syndrome. [53] [92] [118] [116]
In the study by Kato et al [118], out of 15 patients, carcinoid syndrome was present in 2 patients, but serotonin secretion was seen in 10 patients.
A metastatic spread was reported in 12% to 20% of the patients with a primary NET of the testis [47] [91] [116]] [118]
The size of the tumour of the testis [91] [92] [108] along with carcinoid syndrome [91] [92] had been suspected to be possible risk factors for the metastatic spread.
Their own search of the literature revealed metastases in 9 out of 51 patients. The age range indicated for 6 patients with metastases was between 25 years and 78 years with an average age of 59 years. Thus, patients with metastases had a higher average age than patients without metastases of approximately 45 years.
Metastases may occur even several years following the orchidectomy due to the primary NET of the testis [119]
In some reports [for example, [53] [120], the good prognosis of the patients with a primary NET of the testis was mentioned, but this did not apply for the minority of the patients with metastatic spread.
The current case report relates to a young patient with a primary NET of the testis, presented was a special case in several aspects.Penke [115] reported a patient who was 19 years old, and who weighed 70 kilograms, with a height of 180 cm, when he reported pain in his right testis and hypogastrium in September 2012. He had not suffered from any other discomfort: neither fever, night sweat, loss of weight nor signs of bleeding or flush symptoms, diarrhoea, or vertigo. He did not have any known history of cancer diseases in his family. In view of the pain and the clear testicular swelling, a dextral orchidectomy was undertaken. The histology revealed a NET of the testis without a teratoid tumour with angioinvasion and infiltration of the peri-orchitic fatty tissue and periorchium. It was possible to detect chromogranin A and synaptophysin within the tumour tissue. The Ki-67 index was 10% and had revealed a histological grading of G2. Serotonin was formed by the tumour tissue. The histological reference result by the Institute of Pathology at the University Clinic of Heidelberg confirmed the diagnosis of a NET of the testis (Ki-67 approx. 5%). Based upon this result, a comprehensive diagnosis in the tumour staging as well as evidence or exclusion of a primary tumour in other locations was undertaken between October 2012 and December 2012: among others, thoracic/abdominal CT, DOTATOC-PET/CT, and cardiac MRI. The thoracic/abdominal CT resulted in the finding of a solitary subpleural consolidation dorsal medial on the left (approx. size 1.7 cm), and two suspicious lymph nodes within the epigastrium (of approximate. size 1.9 cm). The DOTATOC-PET/CT revealed the suspected somatostatin receptor-positive lymph node metastases, retroperitoneal para-aortic bilateral osseous metastases in the area of the sacrum (sacral vertebra), as well as an intramyocardial metastasis. The cardiac MRI supported the suspected intramyocardial masses or metastasis (6 × 4 mm) in the lateral wall of the left ventricle at the top of the heart. The patient was diagnosed as having a functional open primary NET of the dextral testis (Ki-67: 5-10%; G2) with serotonin secretion without hormone-induced clinical symptoms. According to the TNM staging for germinal tumours, the NET was classified as follows: pT2 pN1 pM1b. In addition, the patient was found to have lymph node metastases in the retroperitoneal, and para-aortal lymph nodes, osseous metastases within the second sacral vertebra on the left, and mediastinal metastatic spread as well as myocardial infiltration. In view of the patient's young age, the Interdisciplinary Tumour Board decided to implement a maximally intensive therapeutic concept with the target to achieve a long-term remission by a comprehensive tumour debulking. In December 2012, a radical retroperitoneal lymphadenectomy including appendicectomy was undertaken. The histological examination revealed lymph node metastases of the NET of the testis in 2 of the 12 examined inter-aortocaval lymph nodes with regard to other lymph nodes and the appendix without any tumours. In January and February 2013, the patient was treated with radiation to the sacrum, with a total dosage of 50 Gy in 10 fractions, to treat the osseous metastases. In February 2013, the DOTATOC-PET/CT still showed the osseous metastases in the second sacral vertebra and the suspected intramyocardial metastasis in the lateral wall of the left ventricle. In March 2013, the patient agreed to undergo an exploratory thoracotomy. After prior marking of the tumour with Tc99-DOTATOC, several other myocardial tumour foci with an infiltration of both ventricles were detected under the microscope in addition to the known lesions (PET/CT, cardiac MRI) during the surgery. The intraoperative frozen section analysis revealed NET cells in the biopsies of the suspected metastases. It was not possible to perform a radical curative resection of this comprehensive cardiac metastatic spread. The patient was notified about the results, and due to the impossible curative treatment and the suspected somatostatin receptor within the metastases, the National Centrum for Tumour Diseases in Heidelberg suggested an antiproliferative therapy with the somatostatin analogue lanreotide (lanreotide autogel; 60 mg every 4 weeks). This therapy was commenced in April 2013. AS per August 2013, the regular follow-up with the tumour marker DOTATOC-PET/CT and cardiac MRI had led to a stable disease and wellbeing of the patient. Penke [115] made the ensuing educative summative discussions:
A 19-year-old patient with testicular pain was diagnosed with a histologically very rare NET of the testis. The comprehensive diagnosis revealed lymph node, cardiac, and (suspected) osseous metastases.
It was possible to remove the lymph node metastases through surgery and to radiate the suspected bone metastases in a fractionated way.
An exploratory thoracotomy had shown a comprehensive, not curatively resectable cardiac metastatic spread.
The patient received palliative antiproliferative monthly therapy with lanreotide. After 4 months of therapy, a stable disease situation could be observed.
The presented patient was a special case in the scope of this very rare disease under several aspects.
Within the literature, a maximum of 10 patients under 20 years of age with a primary NET of the testis had been reported. Thus, the patient presented in the report being read was one of the very first with a metastasizing primary NET of the testis at such a young age. Even when regarding bone or cardiac metastases in a primary NET of the testis, only single reports were available. In 1991, metastases in the neck and thoracic vertebrae were reported in a 53-year-old Japanese patient [121], and in 1981, cardiac metastases were reported in a 76-year-old United States of America patient [122].
The commonest symptom of primary NETs of the testis is unpainful or almost unpainful testicular swelling; in the case series of Wang et al. [53], this was the case in 15 out of 24 patients with corresponding information. In 6 out of the 24 patients, the tumour was accidentally detected. Due to this very rare primary NET of the testis, a primary tumour resection was required regarding the histological evidence of a NET in the testis. [123] [124]
A metastasizing NET of the testis that cannot be curatively resected was treated with medication. In this regard, chemotherapy had been proven to be ineffective in most cases, which had already been seen in the case of a 77-year-old Japanese patient who had multiple pulmonary and retroperitoneal lymph node metastases in 1997[125]. In contrast, somatostatin analogues such as octreotide and lanreotide in case of a NET have an antiproliferative effect on the primary tumour and on the metastases. This was also the result of a meta-analysis including 28 trials on the tumour effect of octreotide or lanreotide in case of a NET at different sites. [126] Penke [115] concluded that it is possible to administer lanreotide autogel to control the growth of the tumour in a young patient with a metastasizing primary NET of the testis and with impossible surgical treatment of the metastases. Ghorefi et al. [127] stated the following:
Testicular neuroendocrine tumours, which are commonly called carcinoid, are extremely rare and account for less than 1% of all testicular neoplasms.
The commonest type of neuroendocrine tumour of the testis is primary carcinoid followed by testicular metastasis from another primary and rarely carcinoid within a testicular teratoma.
Up to the time of publication of their article in 2020, less than 25 cases of carcinoid associated teratomas had been reported in the literature. Ghoreifi et al. [127] reported a unique case of testicular carcinoid tumour arising in association with mature teratoma in a patient with contralateral classic seminomaOtto et al. [128] reviewed previous reports of carcinoid (an endocrine tumour mostly of the gastrointestinal tract) tumours of the testis. Otto et al. [] stated the following:
Carcinoid tumours of the testis are rare and can be divided into primary carcinoid (group 1), testicular metastasis from another location (group 2) and carcinoid within a testicular teratoma (group 3).
A case of testicular carcinoid within their clinic had prompted them to review previous reports; all the cases that were found were assessed for patient and tumour characteristics, diagnostic tools used, treatment and prognosis.Otto et al. [128] summarized the results as follows:
In all, 62 cases were assessed and divided into groups: 1 (44 patients), 2 (six) and 3 (12), respectively.
Seven patients in group 1 developed metastases.
A wide variety of diagnostic tools was used to search for other tumour sites.
All patients were treated with the undertaking of orchidectomy. Three patients with a primary carcinoid were treated with adjuvant chemotherapy (two) or radiotherapy (one), with unknown results.
All but one of the nine patients who died were known to have metastasis, either from a primary testicular carcinoid or testicular metastases from an intestinal carcinoid.Otto et al. [128] made the following conclusions:
When a testicular carcinoid tumour is discovered, other tumour sites should be excluded.
The most useful diagnostic tools for this purpose seem to be urinary 5-hydroxyindoleacetic acid measurement, somatostatin receptor scintigraphy, computed tomography and video-capsule endoscopy.
Localized testicular carcinoid tumours have an excellent prognosis after orchidectomy.Wang et al. [129] reported a case of a primary carcinoid tumour of the testis in a 35-year-old man as an incidental finding. The patient underwent ultrasound of testis which demonstrated a 1.1 cm hypoechoic/isoechoic mass with some calcification within his left testicle. The pathology examination of his radical orchiectomy specimen demonstrated a pure carcinoid tumour, with the adjacent coarse calcification. Fluorescence in situ hybridization showed 35 % of the tumour cells had one additional chromosome 12p11.21 signal. Wang et al. [129] stated that their reported case adds to the rare reports in the literature of a primary pure carcinoid tumour of the testis, and provides additional insight into the radiological and pathological correlation of this disease.Nistal and Paniagua [130] reported a 51-year-old man who had presented with metastasis of a small cell carcinoma of unknown origin in a right inguinal lymph node. His clinical and laboratory studies failed to locate the primary tumour. After three years, a swelling appeared within his right testis, which was removed. Histological examination of the testicular specimen revealed a proliferation of small tumour cells forming irregular masses or nests that occupied most of the testicular parenchyma. At the periphery of the testicular parenchyma a few seminiferous tubules could be seen, showing a low and incomplete seminiferous epithelium and numerous tumour cells in the lumen. Most of the tumour cells showed a euchromatic nucleus with small nucleoli and scanty cytoplasm. Among these cells, larger bi-nucleate or trinucleate cells as well as small cells with pycnotic nuclei were also seen. Mitoses were numerous. Electron microscopy of the specimen revealed some tumour cells with 80 to 100 nm vesicles containing electron-dense granules. Some cells had displayed dendrite-like prolongations with numerous intermediate filaments and electron-dense vesicles. Nistal and Paniagua. [130] conclude that the features of the tumour were compatible with a primary neuroectodermal tumour of the testis.Luviano-García et al. [131] stated the following:
Primary neuroendocrine carcinoid testicular tumours are an uncommon histological subtype representing less than 1% of testicular neoplasms, clinically presenting as an indolent or painful testicular mass.
Immunohistochemistry is the cornerstone for definitive diagnosis, being chromogranin, synaptophysin and cytokeratin the most reliable markers.
Radical orchidectomy is the treatment of choice. Luviano-García et al. [131] reported a 31-year-old man with a 2-month history of right testis palpable mass, which was indolent with no other symptoms. He had Doppler ultrasound scan which was reported as showing a solid mass that measured 2.1 cm x 1.7 cm with a mixed pattern within the upper pole of his right testis and enhanced vascularity The Pathology Department report of his orchidectomy specimen revealed a small cell neoplasia with an insular pattern and microfollicular/rosette formations. Necrotic patches were observed in 20% of the reviewed material. The small cells were characterized by eosinophilic cytoplasm and nuclei with granular chromatin. The neoplasia showed lympho-vascular invasion and negative margins. Regarding immunohistochemistry staining results, chromogranin, cytokeratin, and synaptophysin, stains were positive. Only 3% of tumour cells were positive for ki-67. The former described a well-differentiated carcinoid tumour with a non-teratoma component and no germ cell elements. The reported patient was under surveillance by the Oncology and Gastroenterology Departments. A primary extra-testicular carcinoid tumour metastasis was excluded. Six months after surgery, his active surveillance showed no tumour activity. Luviano-García et al. [131] made the following conclusions:
Primary neuroendocrine carcinoid testicular tumours represent an uncommon subtype of low malignant potential neoplasia.
Definitive diagnosis depends upon immunohistochemistry features after radical orchidectomy.
The prognosis depends upon tumour size and the presence of metastasis. Abou Zahr et al. [132] stated the following:
Primary carcinoid tumour of testis is a rare type of testicular cancer accounting for less than 1% of all testicular tumours.
It is associated with teratoma components [91] [133] [47] and could occur in all ages with mean age of onset ranging between 45 and 50. [91] [133]
Among all reported cases, 12% are associated with carcinoid syndrome which is correlated with increased metastatic potential [91].
Macroscopically, carcinoid tumours are well circumscribed with calcifications seen in 10% of cases. [91]
They are arranged in solid nests and acini of cells in a fibrous to hyalinized stroma.
Cells consist of eosinophilic, granular cytoplasm and round nuclei with a punctate chromatin pattern.
Vascular invasion is seen in 20% of testicular carcinoid tumours but is not associated with increased malignant potential [91] [134].
Cells might rarely adopt a trabecular pattern; they stain positive for substance P, serotonin, chromogranin, synaptophysin, and cytokeratin [51] [91] [135] but stain negative for OCT4, CD30, CD117, TTF-1, and CDX-2 [81] OCT4 is a sensitive and specific marker for diagnosing seminoma, dysgerminoma, germinoma, and embryonal carcinoma [47]. CD117 and CD30 are useful in distinguishing embryonal carcinoma and seminoma, dysgerminoma, and germinoma [136].
TTF1 is a homeodomain-containing nuclear transcription protein of the NK2 homeobox (NKX2) gene family which was reported to stain positively in 53% of gastrointestinal (GI) tract carcinomas. CDX-2, caudal type homeobox 2, is used to identify tumours originating from the upper GI tract [137].
Differentiation between primary and metastatic carcinoid tumours of testis is of utmost importance for it correlates with the prognosis.
The presence of teratoma elements in the tumour indicates a primary origin whereas bilateral involvement, multifocal tumours, or extra-testicular extension is indicator of metastatic origin.
Tumour size and the presence of carcinoid syndrome are the two important predictors for the development of metastasis which might reach 16% roughly and hence the need for follow-up.
Primary tumours have good prognosis and are usually cured by orchiectomy. [47] [91]
Follow-up consists of radiology imaging (CT or Magnetic Resonance Imaging) and urinary 5-hydroxyindoleacetic acid (5-HIAA) every three to four months in the first year.
If asymptomatic and complete macroscopic resection was achieved, watchful surveillance can be adopted with yearly urinary 5-HIAA level and radiology imaging only when level is positive [138]
They had presented the case of a primary carcinoid tumour of the testis with a long follow-upAbou Zahr et al. [132] reported a 36-year-old male patient who had manifested with painless enlargement of his right scrotum of a-few-weeks duration. His history did not reveal any trauma or previous infections. His clinical examination demonstrated diffuse right scrotal nontender and a firm mass with no palpable inguinal lymph nodes. He had ultrasound scan which demonstrated an iso-echogenic solid mass that measured 4.2 cm x2.8. cm with two calcified foci at the apex posteriorly, the largest measuring 6.7 mm x7.6 mm. The patient underwent right radical orchiectomy. He had Chest abdomen pelvis Computed Tomography (CT) scan which was negative for metastasis. The patient had been followed up for 7 years and he did not develop any recurrence on his yearly follow-up CT scan and urinary 5-HIAA. Histopathology examination of the orchidectomy specimen showed, macroscopically, parenchyma of the testis partially occupied by a solid, well circumscribed, nonencapsulated mass measuring 5 cm x 4 cm x 3.5cm with a homogenous tan grey to “creamy” colour. Grossly visible areas of haemorrhage and necrosis were absent. Within the centre of the mass, there was a 0.4 cm calcified nodule. The tumour was grossly separated from the tunica albuginea and from testicular hilum by a grossly unremarkable, light brown, spongy soft testicular parenchyma. Microscopically, the neoplasm is composed of cohesive, uniform, cuboidal cells with minimal cytoplasm, arranged in nests and cribriform structures of variable size and shape, separated by moderate amount of fibrotic dense stroma. At the periphery of the nodule, cord-like and trabecular growth patterns were noticed. Confluent as well as individual cell necrosis was absent. The tumour cells exhibited a uniform, round nuclei with regular contour and a granular chromatin. Prominent nucleoli were absent. The cytoplasm was pale, mild, and inconspicuous. Nuclear grooves were absent. Mitotic figures were absent to extremely rare (2 per 10 HPF). The tumour cells stained positive with cytokeratin (clones AE1/AE3), synaptophysin, chromogranin A, EMA, and CDX-2 but the tumour cells stained negatively for CD99, TTF-1, CK7, CK20, and CEA. Cohesive cuboidal cells with minimal amount of cytoplasm, round uniform nuclei with regular contour, finely granular chromatin, no nucleoli, and no mitotic activity (H&E; 200x). Abou Zahr et al. [132] made the ensuing discussions:
The term “carcinoid” was used to indicate a group of tumours in the small intestine that arose from neuroendocrine (NE) cells that had low malignant potential [139].
There is still debate regarding the carcinogenesis of primary testicular carcinoid tumours since the presence of NE cells in the testis had not been yet described with hypotheses postulating the origin to be the same precursor cell as Leydig cells, while others attributing it to a chromosomal abnormality. [81] [87]
Testicular carcinoids most commonly manifest as a scrotal mass that could be indolent or painful in nature.
Wang et al. [53] reported 29 primary testicular carcinoid cases and they reported metastases in none of the 20 carcinoid cases with typical features and in 1 of 4 cases with atypical morphology [53]. Metastatic sites were the para-aortic lymph nodes, lungs, vertebrae, retroperitoneum, skin, and skeletal muscle [53].
Stroosma et al. reported metastases in 15.9% of cases of primary testicular lesions with no distinguishing [47]
Carcinoid tumours occur in all age groups. According to Wang et al. the diagnosis is based upon the histopathology when there are no carcinoid symptoms or signs of metastasis. [53]
Ten percent (10%) of carcinoid syndrome cases may metastasize to liver or lungs and manifest by erythema on upper torso and face due to the secretion of vasoactive amines, abdominal pain, diarrhoea, cramps, and attacks of asthma due to the activation of vasoactive amines like serotonin [89]
Histopathology assessment has great value in the classification of carcinoid tumours. Well-differentiated neoplastic cells have mitotic figures less than 2 per 10 HPF with mild cellular atypia. Moderately differentiated carcinoid tumours are characterized by necrosis and moderate cellular atypia and necrosis with mitotic activity more than 3 per 10 HPF and have metastatic potential [53].
Histopathologic assessment of their patient was well-differentiated carcinoid tumour.
It was reported that primary testicular carcinoids are CDX-2 negative on immunocytohistochemistry staining [81]; nevertheless, a study by Lee et al. had suggested that a primary testicular tumour can stain positive with CDX-2 and hence be misleading when dealing with a metastatic tumour with unknown primary which can highly be due to an occult testicular malignancy rather than of GI origin [140].
In their case, CDX-2 staining was positive; however, no GI origin was found which goes in favour of occult testicular malignancy. In a gross appearance, testicular carcinoids have been found to measure between 1 and 9.5 cm and are solid, lobulated with yellow to dark-tan and may or may not have calcifications. [135]
Histologically, they are trabeculated with or without central necrosis depending on the size [140]. Grade 1 tumour cells are monomorphic with round nuclei, finely dispersed chromatin, acidophilic finely granular cytoplasm, low mitotic activity. With less than <2>
Histologically, their presented case had demonstrated no atypical features.
The mainstay treatment for a localized carcinoid tumour of the testis is by focal excision or radical orchiectomy, even though chemotherapy and adjuvant radiotherapy had been undertaken in metastatic cases [53].
Majority of t carcinoid tumours are benign and have portended a good prognosis; nevertheless, the long-term prognosis is dependent upon the histopathology- grade.
Wang et al. in their study had shown that after a mean follow-up of 52.7 months all cases of patients with typical features upon histology examination had no recurrence or metastases, whereas 25% of those who had atypical histology developed metastases [53].
Sasaki et al. [141] had shown metastasis 6 years after orchidectomy which indicated the need for long-term follow-up of patients [141]. Abou Zahr et al. [132] made the following conclusions:
Carcinoid tumours of the testis are uncommon, different in manifestation, and affecting all age groups.
Primary carcinoid tumours have good prognosis but the metastatic potential exists and may be delayed; thus, a long-term follow-up is warranted. Petrossian et al. [142] stated the following:
It has been documented that Carcinoid tumours are the most commonly encountered small bowel malignancy and are believed to arise from neuroendocrine cells [143]
Carcinoid tumours are classically found at the tip of the vermiform appendix or the terminal ileum although they have also been described to occur in the lungs, pancreas, rectum, and genitourinary tract. [144]
Carcinoid tumours of the testis are a rare entity comprising less than one percent of all testis tumours [92].
Carcinoid tumours of the testis could arise as a metastasis from an extra-testicular primary, as a component of a teratoma, or as a primary tumour, with primary tumour being most common. [47]
In their report, they had described a case of a primary carcinoid tumour of the testis and literature review, with focus on presentation, diagnostic and pathologic evaluation, surgical management, and follow-up.Petrossian et al. [142] reported a patient GD, who was a 78-year-old Caucasian man referred with a four-month history of an enlarging, minimally tender left scrotal mass. He denied any weight loss, trauma, haematuria, or systemic symptoms. His clinical examination demonstrated an indurated left testis which was consistent with a mass. The results of his urinalysis and microscopy were negative for infection. He had ultrasound scan of his testes and scrotal contents which revealed a 3.4 cm × 3.0 cm well-circumscribed, hyperaemic, cystic, and solid lesion of mixed echogenicity within his left testis which was highly suspicious for malignancy (see figure 12). The results of his serum tumour markers and staging CT scan were normal and had demonstrated no evidence of metastasis. The patient underwent an uncomplicated left radical orchiectomy.
Figure 12: Scrotal ultrasound of left testis (sagittal and transverse views). Note the cystic and solid lesion of mixed echogenicity. Reproduced from [142] under the Creative Commons Attribution License.
Grossly, the orchidectomy specimen showed a 3.4 cm well-circumscribed, yellow-tan mass with haemorrhagic areas (see figure 13). The lesion was noted to be predominantly solid with an ill-defined cystic area filled with straw coloured fluid. Histologically, the tumour had classic features of a carcinoid. The tumour cells were noted to be arranged in insular, glandular, and trabecular patterns in a background of fibrous stroma. The cells had abundant granular, eosinophilic cytoplasm and round to oval nuclei with a “salt and pepper” chromatin pattern. No teratoma elements or other germ cell components were identified. A thorough sampling of the adjacent grossly normal testicular parenchyma had shown only atrophic seminiferous tubules with no evidence of intratubular germ cell neoplasia. Immunostaining examinations for cytokeratin AE1/AE3 and neuroendocrine markers (chromogranin, synaptophysin, and CD56) were strongly positive supporting the diagnosis of a carcinoid tumour (see figure 14). The specimen did not appear to have any aggressive features such as multifocality, bilaterality, extra-testicular extension, or evidence of vascular invasion. In addition, given its smaller size and lack of systemic symptoms, the risk of metastasis was determined to be low.
Figure 13: Specimen photograph of left testis showing a 3.4 cm well-circumscribed, yellow-tan mass with haemorrhagic areas; Reproduced from [142] Under Creative Commons Attribution License.
Figure 14: Positive synaptophysin and chromogranin staining of left testis carcinoid tumour. Reproduced from [142] Under the Creative Commons Attribution license.
Given the lack of a defined protocol surrounding this entity, and its likely indolent course, the collective decision made proceeds with close observation consisting of periodic cross-sectional imaging. No biochemical follow-up is planned given absence of carcinoid symptoms on presentation and low-risk pathological features. At the time of this writing (12-month follow-up), the patient was without evidence of recurrence. Petrossian et al. [142] made the ensuing summative discussions:
The first reported case of primary testicular carcinoid was described by Simon et al. in 1954, with more than 50 cases reported at the time of this writing. [47] [83] [97] [116] [119] [139] [145] [146] [147].
Even though less frequent, additional cases of metastatic carcinoid to testis and carcinoid tumour associated with teratoma had been reported.
The origin of testicular carcinoid is unclear.
Carcinoid tumours of the testis might may arise as a result of differentiation of pluripotential germ cells to argentaffin-like cells or due to the development of a teratoma without any other teratomatous elements [148]. This postulate had been further reinforced by FISH analysis performed by Abbosh et al. which demonstrated the presence of classic genetic alterations that characterize germ cell tumours in these cells with carcinoid lesions: 12 p isochromosomy and overrepresentation [81].
It has been documented that patient who have testicular carcinoids are usually asymptomatic but they may have testicular tenderness, hydrocele, or cryptorchidism on exam. Rarely, these individuals may experience carcinoid syndrome (episodic flushing, diarrhoea, wheezing, and right-sided heart murmurs) as a result of the production of bioactive compounds such as serotonin, histamine, bradykinin, and prostaglandins [149].
Elevated serotonin levels and levels of its metabolite and 5-hydroxyindoleacetic acid (5-HIAA) are present in the blood and urine of those patients with this syndrome [150]. Traditionally, liver or lung metastases should be present to result in the manifestation of carcinoid syndrome [83].
In majority of cases, the standard work-up for a testicular tumour had already been undertaken preoperatively which may include serum tumour markers, and cross-sectional imaging to assess for lymphadenopathy. Nevertheless, given the likelihood of an extra-testicular primary, dedicated radiology imaging of the lungs, abdomen and pelvis should be conducted with either plain films, chest/abdomen/pelvic CT, octreotide scintigraphy, video endoscopy, or a small bowel follow through [47].
Somatostatin receptor scintigraphy using indium-111 labelled octreotide is superior to CT scan in the localization of the primary tumour site and has a 96% sensitivity for detecting metastases. This unique radiology imaging modality could identify about two-thirds of the primary and metastatic tumours. Octreotide binds to type 2 somatostatin receptors which are expressed by most carcinoid cells [83].
It has been iterated that Radical orchidectomy has been considered the treatment of choice as chemotherapy and radiotherapy are known to have little effect on carcinoid tumours of other origin [110].
Pathologically, the lesions are described as solid yellow-tan in appearance with an exceedingly firm texture due to striking desmoplasia which is characteristically present.
Histologically, the neoplastic cells can form discrete islands, trabeculae, strands, glands, or undifferentiated sheets.
Immunohistochemical studies show reactivity to antibodies to cytokeratin AE1 and AE3, chromogranin-A, neuron specific enolase, synaptophysin, and CD56 [149].
Much like pheochromocytomas, the lesions malignant potential cannot be predicted by its histopathology appearance [151].
Regardless, extensive testicular sampling should be undertaken to discover intratubular germ cell neoplasia, a minute teratoma, and/or evidence of a scar representing a burnt out or regressed germ cell component.
Only once these steps had been undertaken could one comfortably confirm a tumour as a primary carcinoid [152].
A review of cases that had been reported to date had indicated that most cases of primary testicular carcinoids have an excellent prognosis following the undertaking of orchidectomy.
In view of the scarcity of this entity, data regarding prognostic indicators is lacking. Nevertheless, literature review had suggested that tumour size, invasion, and evidence of the carcinoid syndrome are associated with a worse prognosis and the development of metastases [91].
Stroosma and Delaere had identified a mean tumour size of 70 mm versus 36 mm in patients with and without metastasis, respectively [47].
Similar findings were seen in gastrointestinal carcinoid tumours even though, once again, the understanding of predictive features had been complicated by the scarcity of these tumours [47]. The study of gastrointestinal carcinoid tumours had seen advances in genetic, molecular, and cellular sciences which may enable the development of tumour markers to provide such predictive information. [153] Similar techniques might evolve in order to improve the understanding of testicular carcinoid; nevertheless, at the present time such markers are lacking.
The overall incidence of metastases is only 11% as was seen in the literature, involving spread to lymph nodes, liver, skin, and the skeletal system. [92] [152]
The vast majority of metastases do tend to be identified at the time of the initial diagnosis and are seen in patients who have high-risk pathologic features [47].
Rare cases of delayed metastases had been reported; nevertheless, making surveillance necessary. [119] [154]
The method of surveillance has been poorly defined given absence of well-defined protocols.
Literature review had suggested significant variability to surveillance used in previously reported cases (chest radiography, CT, or 5-HIAA measurement), with many authors failing to provide information about follow-up protocol they had used. Nonetheless, Petrossian et al. [142] in addition iterated the ensuing:
It was their belief that it is reasonable to undertake annual clinical examination coupled with abdominal and lung radiology imaging as an initial surveillance protocol.
Nevertheless, until further data regarding optimal surveillance is available, they also believe that the decision regarding the extent of surveillance should be made with the patient following an informed discussion.
Serum levels of 5-HIAA or chromogranin A may be utilized; however, this does appear to be of most benefit in patients who manifest initially with carcinoid syndrome.
Conclusions
Neuroendocrine tumours of the testis are rare tumours which clinicians all over the world should be aware so that they can have a high-index of suspicion for the tumour to enable them establish a prompt diagnosis of the tumour.
It is important for clinicians to be able differentiate between primary testicular neuroendocrine tumour (NET) and metastasis of NET to the testis, in view of the fact that both primary and metastatic neuroendocrine tumour of the testis are possible, and consequences of a missed primary tumour in a patient who has metastatic NET are significant.
Clinicians should strongly consider their undertaking of esophago-gastroduodenoscopy and colonoscopy in order to complete a staging work up in any patient who has NETs of his testis.
Patients who have NET of testis would need to undergo follow-up assessments for many years even following their successful treatment in view of the fact that NET has the potential to metastasize even after their initial treatment of curative intent.
Wang Z, Yang Z, Wang W, Chen LD, Huang Y, Li W, Liu JY, Xie XY, Lu MD, Lin MX. (2015). Diagnosis of Testicular Adrenal Rest Tumors on Ultrasound: A Retrospective Study of 15 Cases Report. Medicine (Baltimore). 94(36):1471. View
at Publisher |
View
at Google Scholar
Speiser PW, White PC. Congenital adrenal hyperplasia. N Engl J Med. (2003). Aug 21;349(8):776-788. doi: 10.1056/NEJMra021561. PMID: 12930931. View
at Publisher |
View
at Google Scholar
Claahsen-van der Grinten HL, Dehzad F, Kamphuis-van Ulzen K, de Korte CL. (2014). Increased prevalence of testicular adrenal rest tumours during adolescence in congenital adrenal hyperplasia. Horm Res Paediatr.;82(4):238-244. View
at Publisher |
View
at Google Scholar
Aycan Z, Bas VN, Cetinkaya S, Yilmaz Agladioglu S, Tiryaki T. (2013). Prevalence and long-term follow-up outcomes of testicular adrenal rest tumours in children and adolescent males with congenital adrenal hyperplasia. Clin Endocrinol (Oxf). 78(5):667-672. View
at Publisher |
View
at Google Scholar
Bouman A, Hulsbergen-van de Kaa C, Claahsen-van der Grinten HL. Prevalence of testicular adrenal rest tissue in neonates. Horm Res Paediatr. 2011 Feb;75(2):90-93. View
at Publisher |
View
at Google Scholar
Dogra V, Nathan J, Bhatt S. (2004). Sonographic appearance of testicular adrenal rest tissue in congenital adrenal hyperplasia. J Ultrasound Med. 23(7):979-891. View
at Publisher |
View
at Google Scholar
Avila NA, Premkumar A, Shawker TH, Jones JV, Laue L, Cutler GB Jr. Testicular adrenal rest tissue in congenital adrenal hyperplasia: findings at Gray-scale and color Doppler US. Radiology. 1996 Jan;198(1):99-104. View
at Publisher |
View
at Google Scholar
Stikkelbroeck NM, Suliman HM, Otten BJ, Hermus AR, Blickman JG, Jager GJ. Testicular adrenal rest tumours in post pubertal males with congenital adrenal hyperplasia: sonographic and MR features. Eur Radiol. 2003 Jul;13(7):1597-1603. View
at Publisher |
View
at Google Scholar
Nagamine WH, Mehta SV, Vade A. (2005). Testicular adrenal rest tumors in a patient with congenital adrenal hyperplasia: sonographic and magnetic resonance imaging findings. J Ultrasound Med.;24(12):1717-1720. View
at Publisher |
View
at Google Scholar
Wilkins, L., Fleishmann, W., Howard, J.E. et al. (1940) Macrogenitosomia precox associated with hyperplasia of the androgenic tissue of the adrenal and death from corticoadrenal insufficiency. Endocrinology, 26, 385-395. View
at Publisher |
View
at Google Scholar
Stikkelbroeck NM, Otten BJ, Pasic A, Jager GJ, Sweep CG, Noordam K, Hermus AR. (2001). High prevalence of testicular adrenal rest tumors, impaired spermatogenesis, and Leydig cell failure in adolescent and adult males with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 86(12):5721-5728. View
at Publisher |
View
at Google Scholar
İclal Bayhan G, Cetinkaya S, Gökçe Çinar H, Aycan Z. (2010). Testicular Adrenal Rest Tumor in a Patient with 11β-Hydroxylase Deficient Congenital Adrenal Hyperplasia. Journal of Pediatric Endocrinology and Metabolism. 23(7):729-32. View
at Publisher |
View
at Google Scholar
Erdogan S, Ergin M, Cevlik F, Yuksel B, Tuncer R, Tunali N, Polat S. (2006). Testicular adrenal rest hyperplasia due to 21-hydroxylase deficiency: a case report. Endocr Pathol. Spring;17(1):83-7. View
at Publisher |
View
at Google Scholar
Walker BR, Skoog SJ, Winslow BH, Canning DA, Tank ES. (1997). Testis sparing surgery for steroid unresponsive testicular tumors of the adrenogenital syndrome. The Journal of urology. ;157(4):1460-3. View
at Publisher |
View
at Google Scholar
Vanzulli A, DelMaschio A, Paesano P, Braggion F, Livieri C, Angeli E, Tomasi G, Gatti C, Severi F, Chiumello G. (1992). Testicular masses in association with adrenogenital syndrome: US findings. Radiology. 183(2):425-429. View
at Publisher |
View
at Google Scholar
Claahsen-van der Grinten HL, Otten BJ, Hermus AR, Sweep FC, Hulsbergen-van de Kaa CA. (2008). Testicular adrenal rest tumors in patients with congenital adrenal hyperplasia can cause severe testicular damage. Fertility and sterility. 1;89(3):597-601. View
at Publisher |
View
at Google Scholar
Claahsen-van der Grinten HL, Otten BJ, Takahashi S, Meuleman EJ, Hulsbergen-van de Kaa C, Sweep FC, Hermus AR. (2007). Testicular adrenal rest tumors in adult males with congenital adrenal hyperplasia: evaluation of pituitary-gonadal function before and after successful testis-sparing surgery in eight patients. The Journal of Clinical Endocrinology & Metabolism. 1;92(2):612-5. View
at Publisher |
View
at Google Scholar
Cakir ED, Mutlu FS, Eren E, Paşa AO, Sağlam H, Tarim O. (2012). Testicular adrenal rest tumors in patients with congenital adrenal hyperplasia. Journal of clinical research in pediatric endocrinology. 1;4(2):94-100. View
at Publisher |
View
at Google Scholar
Claahsen-van der Grinten HL, Sweep FC, Blickman JG, Hermus AR, Otten BJ. (2007). Prevalence of testicular adrenal rest tumours in male children with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. European journal of endocrinology. 1;157(3):339-44. View
at Publisher |
View
at Google Scholar
Shanklin DR, Richardson AP, Rothstein G. (1963). Testicular hilar nodules in adrenogenital syndrome: the nature of the nodules. American Journal of Diseases of Children. 1;106(3):243-50. View
at Publisher |
View
at Google Scholar
Rich MA, Keating MA. (2000). Leydig cell tumors and tumors associated with congenital adrenal hyperplasia. Urol Clin North Am. 27(3):519-528. View
at Publisher |
View
at Google Scholar
Kamoun M, Feki MM, Sfar MH, Abid M. (2013). Congenital adrenal hyperplasia: Treatment and outcomes. Indian J Endocrinol Metab. 17(1):14-7. View
at Publisher |
View
at Google Scholar
Cabrera MS, Vogiatzi MG, New MI. (2001). Long term outcome in adult males with classic congenital adrenal hyperplasia. J Clin Endocrinol Metab. ;86(7):3070-8. View
at Publisher |
View
at Google Scholar
Finkielstain GP, Kim MS, Sinaii N, Nishitani M, Van Ryzin C, Hill SC, Reynolds JC, Hanna RM, Merke DP. (2012). Clinical characteristics of a cohort of 244 patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab;97(12):4429-38. View
at Publisher |
View
at Google Scholar
DAHL EV, BAHN RC. (1962). Aberrant adrenal contical tissue near the testis in human infants. Am J Pathol. 40(5):587-598. PMID: 13883088; PMCID: PMC1949556. View
at Publisher |
View
at Google Scholar
Sullivan JG, Gohel M, Kinder RB. (2005). Ectopic adrenocortical tissue found at groin exploration in children: incidence in relation to diagnosis, age and sex. BJU Int;95(3):407-410. View
at Publisher |
View
at Google Scholar
Souverijns G, Peene P, Keuleers H, Vanbockrijck M. (2000). Ectopic localisation of adrenal cortex. Eur Radiol.;10(7):1165-1168. View
at Publisher |
View
at Google Scholar
Clark RV, Albertson BD, Munabi A, Cassorla F, Aguilera G, Warren DW, Sherins RJ, Loriaux DL. (1990). Steroidogenic enzyme activities, morphology, and receptor studies of a testicular adrenal rest in a patient with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 70(5):1408-13. View
at Publisher |
View
at Google Scholar
Bercovici JP, Fiet J, Gibault L, Volant A, Abalain JH, Floch HH, Sonnet E, Fournier G. (2005). Testicular adrenal rest tumours in salt wasting congenital adrenal hyperplasia (in vivo and in vitro studies). J Steroid Biochem Mol Biol. 93(1):67-72. View
at Publisher |
View
at Google Scholar
Combes-Moukhovsky ME, Kottler ML, Valensi P, Boudou P, Sibony M, Attali JR. (1994). Gonadal and adrenal catheterization during adrenal suppression and gonadal stimulation in a patient with bilateral testicular tumors and congenital adrenal hyperplasia. J Clin Endocrinol Metab. 79(5):1390-1394 View
at Publisher |
View
at Google Scholar
Blumberg-Tick J, Boudou P, Nahoul K, Schaison G. (1991). Testicular tumors in congenital adrenal hyperplasia: steroid measurements from adrenal and spermatic veins. J Clin Endocrinol Metab. 73(5):1129-1133. View
at Publisher |
View
at Google Scholar
Claahsen-van der Grinten HL, Otten BJ, Stikkelbroeck MM, Sweep FC, Hermus AR. (2009). Testicular adrenal rest tumours in congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab.;23(2):209-20. View
at Publisher |
View
at Google Scholar
Chatelain D, Montel V, Dickes-Coopman A, Chatelain A, Deloof S. (2003). Trophic and steroidogenic effects of water deprivation on the adrenal gland of the adult female rat. Regul Pept. 28;110(3):249-55. View
at Publisher |
View
at Google Scholar
Benvenga S, Smedile G, Lo Giudice F, Trimarchi F. (1993). Testicular adrenal rests: evidence for luteinizing hormone receptors and for distinct types of testicular nodules differing for their autonomization. Eur J Endocrinol.;141(3):231-7. View
at Publisher |
View
at Google Scholar
Murphy H, George C, de Kretser D, Judd S. (2001). Successful treatment with ICSI of infertility caused by azoospermia associated with adrenal rests in the testes: case report. Hum Reprod.;16(2):263-267 View
at Publisher |
View
at Google Scholar
san Miguel Fraile P, Fernández Fernández G, Meijide Rico F, Antón Badiola I, Ortiz-Rey JA, Alvarez Alvarez C, de la Fuente Buceta A. (2003). Hiperplasia de restos adrenales en el testículo: una causa infrecuente de infertilidad masculina [Hyperplasia of adrenal rests in the testicle: a rare cause of male infertility]. Actas Urol Esp. 27(3):234-239. Spanish. View
at Publisher |
View
at Google Scholar
Wang Z, Yang S, Shi H, Du H, Xue L, Wang L, Dong Y, Han A. (2011). Histopathological and immunophenotypic features of testicular tumour of the adrenogenital syndrome. Histopathology;58(7):1013-1018 View
at Publisher |
View
at Google Scholar
Leotta A, Lio SG. (1994). Bilateral interstitial cell tumor of the testis: a report of a case in an adult. Pathologica. ;86(5):557-559. View
at Publisher |
View
at Google Scholar
Heijkoop B, Perera M, Kelly BD, Bolton D. (2021). Metastatic neuroendocrine tumour presenting as a testicular mass. BMJ Case Rep. 4;14(2): 240042. View
at Publisher |
View
at Google Scholar
Hofland J, Kaltsas G, de Herder WW. (2020). Advances in the diagnosis and management of well-differentiated neuroendocrine neoplasms. Endocrine Reviews.;41(2):371-403. View
at Publisher |
View
at Google Scholar
Oronsky B, Ma PC, Morgensztern D, Carter CA. Nothing But NET: (2017). A Review of Neuroendocrine Tumors and Carcinomas. Neoplasia;19(12):991-1002. View
at Publisher |
View
at Google Scholar
Amine MM, Mohamed B, Mourad H, Majed H, Slim C, Mehdi B, Hela M, Nouri R, Rim K, Tahya B, Nabil MM. (2017). Neuroendocrine Testicular Tumors: A Systematic Review and Meta-Analysis. Curr Urol. 10(1):15-25. View
at Publisher |
View
at Google Scholar
Xiao GQ, Birns DR, Warner R R, Selleck W, Unger P D. (2004). Testicular metastasis of primary cecal carcinoid tumor. Annals of DiagnosticPathology 01; 8(2):102–107. View
at Publisher |
View
at Google Scholar
Fucs M, Romero FR, Germanos de Castro M, de Carvalho Fernandes R, Camara-Lopes LH, Cardenuto Perez MD. (2005). Testicular metastasis 10 years after resection of appendiceal carcinoid. Urology;65(3):591. View
at Publisher |
View
at Google Scholar
Lamarca A, Clouston H, Barriuso J, McNamara MG, Frizziero M, Mansoor W, Hubner RA, Manoharan P, O'Dwyer S, Valle JW. (2019). Follow-Up Recommendations after Curative Resection of Well-Differentiated Neuroendocrine Tumours: Review of Current Evidence and Clinical Practice. J Clin Med. 5;8(10):1630. View
at Publisher |
View
at Google Scholar
Hofland J, Kaltsas G, de Herder WW. (2020). Advances in the Diagnosis and Management of Well-Differentiated Neuroendocrine Neoplasms. Endocr Rev. 1;41(2):371–403. View
at Publisher |
View
at Google Scholar
Reyes A, Moran CA, Suster S, Michal M, Dominguez H. (2003). Neuroendocrine carcinomas (carcinoid tumor) of the testis. A clinicopathologic and immunohistochemical study of ten cases. Am J Clin Pathol. 120(2):182-7. View
at Publisher |
View
at Google Scholar
Han X, Yu L, Yang S, Zheng J. Primary neuroendocrine tumor of the testis: a study of clinicopathological features. Int J Clin Exp Pathol. 2014 Mar 15;7(4):1771-17766. PMID: 24817977; PMCID: PMC4014261. View
at Publisher |
View
at Google Scholar
Wang WP, Guo C, Berney DM, Ulbright TM, Hansel DE, Shen R, Ali T, Epstein JI. (2010). Primary carcinoid tumors of the testis: a clinicopathologic study of 29 cases. Am J Surg Pathol. 34(4):519-24. View
at Publisher |
View
at Google Scholar
Mweempwa, A., Tan, A. & Dray, M. Recurrent Merkel cell carcinoma of the testis with unknown primary site: a case report. J Med Case Reports 10, 314 (2016). View
at Publisher |
View
at Google Scholar
Albores-Saavedra J, Batich K, Chable-Montero F, Sagy N, Schwartz AM, Henson DE. (2010). Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population-based study. J Cutan Pathol. 37(1):20-7. View
at Publisher |
View
at Google Scholar
Clarke CA, Robbins HA, Tatalovich Z, Lynch CF, Pawlish KS, Finch JL, Hernandez BY, Fraumeni JF Jr, Madeleine MM, Engels EA. (2015). Risk of merkel cell carcinoma after solid organ transplantation. J Natl Cancer Inst. 8;107(2):382. View
at Publisher |
View
at Google Scholar
Tadmor T, Aviv A, Polliack A. (2011). Merkel cell carcinoma, chronic lymphocytic leukemia and other lymphoproliferative disorders: an old bond with possible new viral ties. Ann Oncol.;22(2):250-6 View
at Publisher |
View
at Google Scholar
Engels EA, Frisch M, Goedert JJ, Biggar RJ, Miller RW. (2011). Merkel cell carcinoma and HIV infection. Lancet. 9;359(9305):497-8. View
at Publisher |
View
at Google Scholar
origin in merkel cell carcinoma? J Skin Cancer. 2012; 2012:680410. doi: 10.1155/2012/680410. Epub 2012 Dec 13. PMID: 23304516; PMCID: PMC3530849. View
at Publisher |
View
at Google Scholar
Zur Hausen A, Rennspiess D, Winnepenninckx V, Speel EJ, Kurz AK. (2013). Early B-cell differentiation in Merkel cell carcinomas: clues to cellular ancestry. Cancer Res. 15;73(16):4982-7. View
at Publisher |
View
at Google Scholar
Feng H, Shuda M, Chang Y, Moore PS. (2008). Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 22;319(5866):1096-100 View
at Publisher |
View
at Google Scholar
Gleason JM, Köhler TS, Monga M. (2006). Merkel cell carcinoma metastatic to testis. Urology. 1; 67(2): 423.13–14. View
at Publisher |
View
at Google Scholar
Ro JY, Ayala AG, Tetu B, Ordonez NG, el-Naggar A, Grignon DJ, Mackay B. (1990). Merkel cell carcinoma metastatic to the testis. Am J Clin Pathol. 94(4):384-9. View
at Publisher |
View
at Google Scholar
Rufini V, Perotti G, Brunetti M, Crescenzi A, Fadda G, Troncone L. (2004). Unsuspected testicular metastases from Merkel cell carcinoma: a case report with therapeutic implications. Am J Clin Oncol.27(6):636-7. View
at Publisher |
View
at Google Scholar
Schwindl B, Meissner A, Giedl J, Klotz T. (2006). Merkel cell carcinoma -- a rarity in the urogenital tract. Onkologie.;29(7):326-8. View
at Publisher |
View
at Google Scholar
Tummala MK, Hausner PF, McGuire WP, Gipson T, Berkman A. (2006). CASE 1. Testis: a sanctuary site in Merkel cell carcinoma. J Clin Oncol. 20;24(6):1008-9. View
at Publisher |
View
at Google Scholar
Whitman EJ, Brassell SA, Rosner IL, Moncur JT. (2007). Merkel cell carcinoma as a solitary metastasis to the testis. J Clin Oncol. 20;25(24):3785-6. View
at Publisher |
View
at Google Scholar
Finklestein JZ, Miller DR, Feusner J, Stram DO, Baum E, Shina DC, Johnson DG, Gyepes MT, Hammond GD. (1994). Treatment of overt isolated testicular relapse in children on therapy for acute lymphoblastic leukemia. A report from the Childrens Cancer Group. Cancer. 1;73(1):219-23. View
at Publisher |
View
at Google Scholar
Locatelli F, Schrappe M, Bernardo ME, Rutella S. (2012). How I treat relapsed childhood acute lymphoblastic leukemia. Blood. 120(14):2807-16. View
at Publisher |
View
at Google Scholar
Leibovitch I, Little JS Jr, Foster RS, Rowland RG, Bihrle R, Donohue JP. Delayed orchiectomy after chemotherapy for metastatic nonseminomatous germ cell tumors. J Urol. 1996 Mar;155(3):952-4. PMID: 8583615. View
at Publisher |
View
at Google Scholar
Aron M, Zhou M. (2011). Merkel cell carcinoma of the genitourinary tract. Arch Pathol Lab Med. Aug;135(8):1067-71. View
at Publisher |
View
at Google Scholar
Byrd-Gloster AL, Khoor A, Glass LF, Messina JL, Whitsett JA, Livingston SK, Cagle PT. Differential expression of thyroid transcription factor 1 in small cell lung carcinoma and Merkel cell tumor. Hum Pathol. 2000 Jan;31(1):58-62. View
at Publisher |
View
at Google Scholar
Lebbe C, Becker JC, Grob JJ, Malvehy J, Del Marmol V, Pehamberger H, Peris K, Saiag P, Middleton MR, Bastholt L, Testori A, Stratigos A, Garbe C; (2015). European Dermatology Forum (EDF), the European Association of Dermato-Oncology (EADO) and the European Organization for Research and Treatment of Cancer (EORTC). Diagnosis and treatment of Merkel Cell Carcinoma. European consensus-based interdisciplinary guideline. Eur J Cancer.;51(16):2396-403. View
at Publisher |
View
at Google Scholar
Fang LC, Lemos B, Douglas J, Iyer J, Nghiem P. (2010). Radiation monotherapy as regional treatment for lymph node-positive Merkel cell carcinoma. Cancer. 1;116(7):1783-90. View
at Publisher |
View
at Google Scholar
Bhatia S, Iyer JG, Storer B, Moshiri A, Parvathaneni U, Byrd DR, Sondak VK, Gershenwald JE, Sober A, Nghiem P. Adjuvant radiation therapy and chemotherapy in Merkel cell carcinoma: Survival analysis of 6,908 cases from the National Cancer Data Base. View
at Publisher |
View
at Google Scholar
Chen MM, Roman SA, Sosa JA, Judson BL. (2015). The role of adjuvant therapy in the management of head and neck merkel cell carcinoma: an analysis of 4815 patients. JAMA Otolaryngol Head Neck Surg. ;141(2):137-41. View
at Publisher |
View
at Google Scholar
Voog E, Biron P, Martin JP, Blay JY. (1999). Chemotherapy for patients with locally advanced or metastatic Merkel cell carcinoma. Cancer. 15;85(12):2589-95. View
at Publisher |
View
at Google Scholar
Andea AA, Coit DG, Amin B, Busam KJ. (2008). Merkel cell carcinoma: histologic features and prognosis. Cancer. 1;113(9):2549-58. View
at Publisher |
View
at Google Scholar
Paulson KG, Iyer JG, Tegeder AR, Thibodeau R, Schelter J, Koba S, Schrama D, Simonson WT, Lemos BD, Byrd DR, Koelle DM, Galloway DA, Leonard JH, Madeleine MM, Argenyi ZB, Disis ML, Becker JC, Cleary MA, Nghiem P. (2011). Transcriptome-wide studies of merkel cell carcinoma and validation of intratumoral CD8+ lymphocyte invasion as an independent predictor of survival. J Clin Oncol. 20;29(12):1539-46. View
at Publisher |
View
at Google Scholar
Sihto H, Kukko H, Koljonen V, Sankila R, Böhling T, Joensuu H. (2009). Clinical factors associated with Merkel cell polyomavirus infection in Merkel cell carcinoma. J Natl Cancer Inst. 1;101(13):938-45 View
at Publisher |
View
at Google Scholar
Abbosh PH, Zhang S, Maclennan GT, Montironi R, Lopez-Beltran A, Rank JP, Baldridge LA, Cheng L. (2008). Germ cell origin of testicular carcinoid tumors. Clin Cancer Res. 1;14(5):1393-1396. View
at Publisher |
View
at Google Scholar
Son HY, Ra SW, Jeong JO, Koh EH, Lee HI, Koh JM, Kim WB, Park JY, Shong YK, Lee KU, Kim GS, Kim MS. (2004). Primary carcinoid tumor of the bilateral testis associated with carcinoid syndrome. Int J Urol.;11(11):1041-1043. View
at Publisher |
View
at Google Scholar
Neely D, Gray S. (2011). Primary carcinoid tumour of the testis. Ulster Med J. May;80(2):79-81. PMID: 22347748; PMCID: PMC3229851. View
at Publisher |
View
at Google Scholar
Reyes A, Moran CA, Suster S, Michal M, Dominguez H. (2003). Neuroendocrine carcinomas (carcinoid tumor) of the testis: a clinicopathologic and immunohistochemical study of ten cases. American journal of clinical pathology. 1;120(2):182-7. View
at Publisher |
View
at Google Scholar
Merino J, Zuluaga A, Gutierrez-Tejero F, Del Mar Serrano M, Ciani S, Nogales FF. (2005). Pure testicular carcinoid associated with intratubular germ cell neoplasia. J Clin Pathol. 58(12):1331-3. View
at Publisher |
View
at Google Scholar
Mai KT, Park PC, Yazdi HM, Carlier M. (2006). Leydig cell origin of testicular carcinoid tumour: immunohistochemical and electron microscopic evidence. Histopathology.;49(5):548-9. View
at Publisher |
View
at Google Scholar
Wolf M, Wunderlich H, Hindermann W, Gajda M, Schreiber G, Schubert J. (2006). Case report: Primary carcinoid tumour of the testicle without metastases in combination with testicular atrophy and testosterone deficiency. Int Urol. Nephrol. ;38(3-4):625–628. View
at Publisher |
View
at Google Scholar
Ramage JK, Davies AH, Ardill J, Bax N, Caplin M, Grossman A, Hawkins R, McNicol AM, Reed N, Sutton R, Thakker R, Aylwin S, Breen D, Britton K, Buchanan K, Corrie P, Gillams A, Lewington V, McCance D, Meeran K, Watkinson A; (2005). UKNETwork for Neuroendocrine Tumours. Guidelines for the management of gastroenteropancreatic neuroendocrine (including carcinoid) tumours. Gut. Jun;54 Suppl 4(4): 1-16. View
at Publisher |
View
at Google Scholar
Zavala-Pompa A, Ro JY, el-Naggar A, Ordóñez NG, Amin MB, Pierce PD, Ayala AG. (1993). Primary carcinoid tumor of testis. Immunohistochemical, ultrastructural, and DNA flow cytometric study of three cases with a review of the literature. Cancer. 1;72(5):1726-32. View
at Publisher |
View
at Google Scholar
Hayashi T, Iida S, Taguchi J, Miyajima J, Matsuo M, Tomiyasu K, Matsuoka K, Noda S. (2001). Primary carcinoid of the testis associated with carcinoid syndrome. Int J Urol. ;8(9):522-4. View
at Publisher |
View
at Google Scholar
Sutherland RS, Wettlaufer JN, Miller GJ. (1992). Primary carcinoid tumor of the testicle: a case report and management schema. J Urol.;148(3):880-2. View
at Publisher |
View
at Google Scholar
Takada H, Iwatsuki S, Itoh Y, Sato S, Hayase M, Yasui T. (2006). Primary pure carcinoid tumour of the testis: A case report and review of the literature. Arch Ital Urol Androl. 5;88(3):245-246. View
at Publisher |
View
at Google Scholar
Lubana SS, Singh N, Chan HC, Heimann D. (2015). Primary neuroendocrine tumor (carcinoid tumor) of the testis: a case report with review of literature. Am J Case Rep. 31; 16:328-32. View
at Publisher |
View
at Google Scholar
Langhans TH. (1867). Ueber einen Drüsenpolyp im ileum. Archiv für pathologische Anatomie und Physiologie und für klinische Medicin.;38(4):559-60. View
at Publisher |
View
at Google Scholar
Simon HB, McDonald JR, Culp OS. (1974). Argentaffin tumor (carcinoid) occurring in a benign cystic teratoma of the testicle. J Urol.;72(5):892-894. View
at Publisher |
View
at Google Scholar
Leake J, Levitt G, Ramani P. Primary carcinoid of the testis in a 10-year-old boy. Histopathology. 1991; 19:373–375. View
at Publisher |
View
at Google Scholar
Ting WH, Hsiao SM, Lin HH, Wei MC. (2014). Primary carcinoid tumor of the ovary arising in a mature cystic teratoma: a case report. Eur J Gynaecol Oncol;35(1):100-102. PMID: 24654475. View
at Publisher |
View
at Google Scholar
Guney N, Sayilgan T, Derin D, Ozcan D. (2009). Primary carcinoid tumor arising in a mature cystic teratoma of the ovary: a case report. Eur J Gynaecol Oncol.;30(2):223-5. View
at Publisher |
View
at Google Scholar
Mazzucchelli R, Morichetti D, Lopez-Beltran A, Cheng L, Scarpelli M, Kirkali Z, Montironi R. (2009). Neuroendocrine tumours of the urinary system and male genital organs: clinical significance. BJU Int. Jun;103(11):1464-70. View
at Publisher |
View
at Google Scholar
Bajetta E, Ferrari L, Martinetti A, Celio L, Procopio G, Artale S, Zilembo N, Di Bartolomeo M, Seregni E, Bombardieri E. (1999). Chromogranin A, neuron specific enolase, carcinoembryonic antigen, and hydroxyindole acetic acid evaluation in patients with neuroendocrine tumors. Cancer. 1;86(5):858-65. View
at Publisher |
View
at Google Scholar
Meijer WG, Kema IP, Volmer M, Willemse PH, de Vries EG. (2000). Discriminating capacity of indole markers in the diagnosis of carcinoid tumors. Clin Chem. 46(10):1588-96. View
at Publisher |
View
at Google Scholar
Pasieka JL. (2002). Regression of metastatic carcinoid tumors with octreotide therapy: two case reports and a review of the literature. J Surg Oncol.;79(3):180-7. View
at Publisher |
View
at Google Scholar
Shimura S, Uchida T, Shitara T, et al (1991). Primary carcinoid tumor of the testis with metastasis to the upper vertebrae. Report of a case (in Japanese). Nihon Hinyokika Gakkai Zasshi 82:1157-1160. View
at Publisher |
View
at Google Scholar
Sullivan JL, Packer JT, Bryant M. (1981). Primary malignant carcinoid of the testis. Arch Pathol Lab Med.;105(10):515-7. View
at Publisher |
View
at Google Scholar
Meijer WG, Kema IP, Volmer M, Willemse PH, de Vries EG. (2000). Discriminating capacity of indole markers in the diagnosis of carcinoid tumors. Clin Chem. 46(10):1588-96. View
at Publisher |
View
at Google Scholar
Fujita K, Wada R, Sakurai T, Sashide K, Fujime M. (2005). Primary carcinoid tumor of the testis with teratoma metastatic to the para-aortic lymph node. Int J Urol.;12(3):328-31. View
at Publisher |
View
at Google Scholar
Blumberg JM, Sedberry S, Kazmi SO. (2005). Bilateral asynchronous metastatic carcinoid tumor of the testis. Urology;65(1):174. View
at Publisher |
View
at Google Scholar
Ballesteros C, Del Portillo Sánchez L, Sánchez Yuste R, Sola Galarza I, Martínez Salamanca JI, Carballido Rodríguez J. (2008). Tumor carcinoide testicular asociado a teratoma: a propósito de un caso [Testicular carcinoid tumor associated with teratoma: with regard to a case]. Actas Urol Esp.;32(4):458-60. View
at Publisher |
View
at Google Scholar
Prunoiu VM, Marincaş AM, Alexandru A, Radu M, Proca TM, Răvaş MM, Brătucu E, Ionescu S. (2009). A Rare Case of a Testicular Teratoma Associated with a Neuroendocrine Tumour. Chirurgia (Bucur). 114(2):300-306. View
at Publisher |
View
at Google Scholar
Darré T, Doukouré B, Kouyaté M, Djiwa T, Kwamé D, Napo-Koura G. (2019). A rare testicular tumor: primary carcinoid tumor. Tumori.;105(6):20-23. View
at Publisher |
View
at Google Scholar
Penke M. Primary neuroendocrine tumor of the testis and osseous, cardiac, and lymph node metastases in a young patient. Case Rep Oncol. 2014 Dec 4;7(3):815-818. View
at Publisher |
View
at Google Scholar
Rathert M, Ubrig B, Atkins DJ, Roth S. (2011). Insuläres Karzinoid des Hodens [Carcinoid tumor of the testis]. Urologe A;50(3):340-2. German. View
at Publisher |
View
at Google Scholar
Alsharif S, Al-Shraim M, Alhadi A, Al-Aown A, Fooshang F, Eid R. (2014). Primary neuroendocrine tumor of the testis. Urol Ann.;6(2):173-5. View
at Publisher |
View
at Google Scholar
Kato N, Motoyama T, Kameda N, Hiruta N, Emura I, Hasegawa G, Murata T, Kimura M, Tsuda H, Ishihara T. (2003). Primary carcinoid tumor of the testis: Immunohistochemical, ultrastructural and FISH analysis with review of the literature. Pathol Int. 53(10):680-5. View
at Publisher |
View
at Google Scholar
Sasaki M, Emura M, Kim U, Shinbo M, Shima T, Suzuki N, Tomioka S, Tanaka M, Nakatsu H, Murakami S, Suzuki Y. (2009). [Primary carcinoid tumor of the testis metastatic to the para-aortic lymph nodes in six years after the first operation: a case report]. Hinyokika Kiyo. ;55(4):233-6. View
at Publisher |
View
at Google Scholar
Han XE, Yu L, Yang S, Zheng J. (2014). Primary neuroendocrine tumor of the testis: a study of clinicopathological features. International Journal of Clinical and Experimental Pathology.;7(4):1771. View
at Publisher |
View
at Google Scholar
Shimura S, Uchida T, Shitara T, Nishimura K, Murayama M, Honda N, Koshiba K. (1991). Primary carcinoid tumor of the testis with metastasis to the upper vertebrae. Report of a case. Nihon Hinyokika Gakkai zasshi. The Japanese Journal of Urology. 1;82(7):1157-60. View
at Publisher |
View
at Google Scholar
Sullivan JL, Packer JT, Bryant M: Primary malignant carcinoid of the testis. Arch Pathol Lab Med (1981); 105:515-517. Sullivan JL, Packer JT, Bryant M. Primary malignant carcinoid of the testis. Arch Pathol Lab Med.;105(10):515-7. PMID: 6895162. View
at Publisher |
View
at Google Scholar
Chang YH, Chuang CK, Wu CT, Ng KF, Liao SK. (2002). Primary carcinoid tumor of the testis: case report. Chang Gung Med J.;25(10):695-9. PMID: 12518782. View
at Publisher |
View
at Google Scholar
Takagi Y, Tanaka J. (1997). [Primary testicular carcinoid tumor with teratoma: a case report]. Hinyokika Kiyo. ;43(2):157-9. View
at Publisher |
View
at Google Scholar
Sidéris L, Dubé P, Rinke A. (2012). Antitumor effects of somatostatin analogs in neuroendocrine tumors. Oncologist.;17(6):747-55. View
at Publisher |
View
at Google Scholar
Ghoreifi A, Liang T, Ballas L, Sherrod A, Djaladat H. (2020). Testicular Carcinoid Tumor in a Patient with Contralateral Classic Seminoma. Urology.;141: 32-34. View
at Publisher |
View
at Google Scholar
Wang G, Xiong W, Tucker T, Tran H, Nappi L, Yuan R. (2001). Primary pure carcinoid tumor of the testis: Radiology, pathology and molecular correlation. Urol Case Rep. 21; 39:101787. View
at Publisher |
View
at Google Scholar
Luviano-García EH, Salas-Ochoa J, Alvizo-López S, Parga-Ramírez JS, Sánchez-Hernández RA, Sierra-Díaz E. Primary neuroendocrine carcinoid testicular tumor: a case report. Bol Col Mex Urol. View
at Publisher |
View
at Google Scholar
Abou Zahr R, Chalhoub K, Mansour E, Chouairy C, Ghazal G, Nohra J. (2008). Primary Carcinoid Tumor of the Testis: A Case Report and Review of the Literature. Case Rep Urol. 3; 2018:3614387. View
at Publisher |
View
at Google Scholar
Kim HJ, Cho MY, Park YN, Kie JH. (1999). Primary carcinoid tumor of the testis: immunohistochemical, ultrastructural and DNA flow cytometric study of two cases. J Korean Med Sci. 14(1):57-62 View
at Publisher |
View
at Google Scholar
Ordóñez NG, Ayala AG, Sneige N, Mackay B. (1982). Immunohistochemical demonstration of multiple neurohormonal polypeptides in a case of pure testicular carcinoid. Am J Clin Pathol.;78(6):860-4. View
at Publisher |
View
at Google Scholar
Teng LH, Lu DH, Xu QZ, Fu YJ, Yang H, He ZL. (2005). [Expression and diagnostic significance of OCT4, CD117 and CD30 in germ cell tumors]. Zhonghua Bing Li Xue Za Zhi. 34(11):711-5. View
at Publisher |
View
at Google Scholar
Lin F, Liu H. (2014). Immunohistochemistry in undifferentiated neoplasm/tumor of uncertain origin. Arch Pathol Lab Med. 138(12):1583-610. View
at Publisher |
View
at Google Scholar
Maroun J, Kocha W, Kvols L, Bjarnason G, Chen E, Germond C, Hanna S, Poitras P, Rayson D, Reid R, Rivera J, Roy A, Shah A, Sideris L, Siu L, Wong R. (2006). Guidelines for the diagnosis and management of carcinoid tumours. Part 1: the gastrointestinal tract. A statement from a Canadian National Carcinoid Expert Group. Curr Oncol.;13(2):67-76. View
at Publisher |
View
at Google Scholar
D'Arrigo L, Costa A, Fraggetta F, Cacciola A, Bonaccorsi A, Savoca F, Aragona F. (2004). Primary carcinoid tumour of the testis: A case-report. Arch Ital Urol Androl. 30;86(3):231-2. View
at Publisher |
View
at Google Scholar
Lee MJ, Vogt AP, Hsiao W, Osunkoya AO. (2012). CDX-2 expression in malignant germ cell tumors of the testes, intratubular germ cell neoplasia, and normal seminiferous tubules. Tumour Biol. 33(6):2185-8 View
at Publisher |
View
at Google Scholar
Ikeri N Z, Nnaji F C, Dawodu O, Igbokwe U, Abdulkareem F B, Banjo A A. (2015). Well, Differentiated Neuroendocrine Carcinoma of the Testis in a Nigerian Male. Journal of Case Reports. 16;5(2):473-8. View
at Publisher |
View
at Google Scholar
Petrossian AA, Habibi J, Rapp DE, Ramnani D. (2015). Primary carcinoid tumor of the testis. Case Rep Urol:687482. View
at Publisher |
View
at Google Scholar
Chang YH, Chuang CK, Wu CT, Ng KF, Liao SK. (2002). Primary carcinoid tumor of the testis: case report. Chang Gung Med J.;25(10):695-9. PMID: 12518782. View
at Publisher |
View
at Google Scholar
Guo X, Yamada S, Wang KY, Shimajiri S, Sasaguri Y. (2010). Case of testicular carcinoid. Journal of UOEH. 1;32(2):213-9. View
at Publisher |
View
at Google Scholar
Epperson JR, Pope NM, Abuzeid MJ. (2013). Rare testicular tumor discovered by assault: an unusual presentation of a primary testicular neuroendocrine tumor grade 2. Case Reports in Pathology. View
at Publisher |
View
at Google Scholar
Liu FF, Zheng JF, Zhou LT, Wang CC, Wang JJ, Shen Q, Yu B, Ma HH, Wang JD, Shi QL. (2014). Primary neuroendocrine tumor of the testis: clinicopathological study of 7 cases. Zhonghua nan ke xue= National Journal of Andrology. 1;20(1):63-7. View
at Publisher |
View
at Google Scholar
Park SB, Kim JK, Cho KS. (2006). Imaging findings of a primary bilateral testicular carcinoid tumor associated with carcinoid syndrome. Journal of ultrasound in medicine.;25(3):413-6. View
at Publisher |
View
at Google Scholar
Goljan, E F. (2011). Rapid Review Pathology 5e ISBN 13: 9780323476683, Elsevier Health Sciences, London, UK, 2nd edition. View
at Publisher |
View
at Google Scholar
Glazier D B, Murphy D F, Barnard N, Cummings K B, Weiss R E. (1999). Primary carcinoid tumour of the testis. BJU International. 83(1): 153-154. View
at Publisher |
View
at Google Scholar
Mazzucchelli R, Morichetti D, Lopez-Beltran A, Cheng L, Scarpelli M, Kirkali Z, Montironi R. (2009). Neuroendocrine tumours of the urinary system and male genital organs: clinical significance. BJU Int.;103(11):1464-70. View
at Publisher |
View
at Google Scholar
Meeker A, Heaphy C. Gastroenteropancreatic endocrine tumors. (2014). Molecular and Cellular Endocrinology. 386(1-2): 101-120. View
at Publisher |
View
at Google Scholar
Hosking DH, Bowman DM, McMorris SL, Ramsey EW. (1981) Primary carcinoid of the testis with metastases. The Journal of Urology. 125(2):255-6. View
at Publisher |
View
at Google Scholar
Dear Editorial Team,
Clinical Medical Reviews and Reports.
My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal.
Best wishes from,
Elena Popa.
Dr Elena Popa
It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.
Dr Nikolaos Andreas Chrysanthakopoulos
Dear Grace Pierce,
Editorial Coordinator of Journal of Clinical Research and Reports,
Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future.
Best wishes from,
Robert W. McGee
Robert W McGee
Dear Grace Pierce,
Editorial Coordinator of Journal of Clinical Research and Reports,
Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal.
Best wishes from,
DR Aibing Rao, Head of R&D
Aibing Rao
I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.
Kashani Mehdi
Dear Mercy Grace,
Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences,
We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage.
We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences!
Best wishes from,
Alla Konstantinovna Politova,