
Research Article | DOI: https://doi.org/10.31579/2690-1897/181
1Department of Medical Biology.
2Department of Pediatric Nephrology and Faculty of Medicine, Çukurova University, 01130 Balcalı-Adana, Turkey.
*Corresponding Author: Osman Demirhan, Department of Medical Biology.
Citation: Osman Demirhan., Aysun Karabay Bayazit, (2024), Mutational and Geographical Distribution of the Ctns Gene in Turkish Patients with Cystinosis, J, Surgical Case Reports and Images, 7(2); DOI:10.31579/2690-1897/181
Copyright: © 2024, Osman Demirhan. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 01 March 2024 | Accepted: 14 March 2024 | Published: 22 March 2024
Keywords: renal tubular acidosis; ocular cystinosis; rickets; growth retardation; mutations; CTNS gene; PCR
Cystinosis is a rare autosomal recessive storage disease that occurs as a result of deficiency of the cystinosine carrier protein caused by mutations in the CTNS gene, which encodes cystinosine, a cysteine carrier in lysosomal membranes. Our study aimed to determine cystinosis gene mutations and their geographical distribution in Turkish pediatric patients with cystinosis. Two brothers and one girl with infantile nephropathic and ocular cystinosis were included in the study. Molecular analyzes included initial multiplex polymerase chain reaction (PCR) to identify a 57 kb deletion in CTNS and analysis by PCR and sequencing of the highly conserved exon-intron splice junctions of all coding regions of the gene. None of the patients had the 57 kb deletion, but a homozygous missense mutation was found. This c.451A>G (p.R151G) mutation has been previously described and is a variant specific to the Turkish population.
Conclusion
Our results show that cystinosin activity is associated with loss of c.451A>G (p.R151G) mutation and mutation in the CTNS gene causes nephropathic and ocular cystinosis. The absence of the 57 kb deletion in the Turkish population and the endemic p.R151G mutation show that the mutation spectrum of the Turkish population is not similar to the European population. However, Turkish patients show allelic heterogeneity in terms of other CTNS gene mutations when compared with Egyptian, Iranian, European and North American cystinosis cases.
Cystinosis is an autosomal recessive disease characterized by the accumulation of the amino acid cystine in the cell. Mutations in the responsible CTNS gene lead to a deficiency of the carrier protein called cystinosin. Cystinosis begins in infancy and leads to multiple organ failure in adulthood, including kidney, eye, thyroid, muscle and pancreas. The prevalence of cystinosis is approximately 1 in 100-200,000 in European and US populations [1], but its frequency in Turkey is unknown. CTNS allelic frequencies have been reported in various populations [2-4] and some of these series have also been observed in Turkish patients [2]. There has previously been a comprehensive study in which CNTS gene mutation screening was conducted in Turkish cystinosis patients [5,6]. However, the mutation spectrum in cystinotic patients in Turkey is still not fully known. More than 140 pathogenic CTNS mutations are known in cystinosis patients around the world. The 57 kb deletion is the most common in approximately 50% of CTNS mutant alleles in patients of Northern European and North American origin [6,7]. However, this mutation is almost absent in Middle Eastern countries [2]. Individuals affected by the disease are generally grouped into three clinical forms depending on the age of onset and the severity of symptoms [8]. In the current study, mutation analysis of the CTNS gene and geographical distributions of mutations were performed in two of three individuals affected by cystinosis in two of nine families living in the Mediterranean Region of Turkey.
2.1 Patients We performed molecular genetic analysis on two of three patients who applied to Çukurova University Faculty of Medicine, Pediatric Nephrology and Metabolic Diseases Unit with complaints of fever, weakness, vomiting, abdominal pain, loss of appetite, polydipsia, polyuria, retardation in the arms and eyes, rickets, and rigidity and growth Open Access Research Article Journal of Surgical Case Reports and Images Osman Demirhan * AUCTORES Globalize your Research J. Surgical Case Reports and Images Copy rights@ Tania Leme da Rocha Martinez, et al, Auctores Publishing LLC – Volume 7(2)-181 www.auctoresonline.org ISSN: 2690-1897 Page 2 of 7 complaints. Cystinosis was diagnosed according to the clinical features and laboratory tests of these patients and their family tree was drawn (Figure 1). Studies were conducted on patients and their relatives after obtaining informed consent, in accordance with the ethics committee rules of Çukurova University, Faculty of Medicine. Blood samples were taken from the patients and their parents. Blood samples for genetic diagnosis were sent to the Centogene Laboratory in Rostock/Germany for diagnostic CTNS mutation analysis, with informed consent. The family members lived in the Mediterranean region of Turkey. The disease was seen in three individuals in two of 9 families (Figure 1). A family of four children whose parents were not consanguineous (III6, III7) had typical clinical features of infantile cystinosis (IV5, IV6). The brothers (PIV5, PIV6) showed early and severe features of the disease (Figure 2). The onset of renal failure was delayed in two boys (PIV5, PIV6); at the 6 month in the older sibling (PIV5) and at the age of 4 in the younger sibling (PIV6).The other patient (PIV10) reached end-stage renal failure at age 15. This patient's parents (III10, III11) was consanguineous, and one of the twin sisters (PIV10) had clinical signs of the disease. The first patient (PIV5) was admitted to the pediatric clinic after the age of 6 months due to a history of weakness, stiffness in the legs and eyes, cough, runny nose and developmental delay. There was no family history of kidney disease, but the father had had heart surgery and the mother had thyroid disease. Currently, the patient is 29 years old and has not developed any other symptoms of disease other than kidney disease (Figure 2). Diffuse corneal crystals were seen in the ophthalmological examination and electron microscope, and it was determined that the disease was chronic renal failure with urinary features. Proteinuria was detected in two affected siblings at the ages of 9 and 10. Kidney biopsy showed focal segmental glomerulosclerosis in both. The growth and development stages of the younger brother (PIV6) were appropriate for his age until he was 6 months old. However, he was admitted to the pediatric clinic due to a history of weakness, loss of appetite, drinking too much water, polyuria, intermittent vomiting and developmental delay from the age of 6 months. He was underweight and had clinical signs of malnutrition, rickets, and moderate dehydration. Biochemical findings in urine and serum tests revealed widespread proximal renal tubular acidosis and renal failure. There were typical clinical findings of rickets. Two patient siblings are now receiving drug treatment, and in addition to the kidneys and eyes, enlargement of the liver and spleen was observed at later ages. Genetic analysis was ordered for sequence analysis of the CTNS gene to confirm the diagnosis. 2.2 Molecular analysis We performed cytogenetic and molecular genetic analysis on two siblings in one of two families with sick children. The other family did not consent for analysis. Blood samples were collected from patients and their parents. Genomic DNA was isolated from the patients' leukocytes according to the standard procedure. Molecular analyses; multiplex PCR was first used to determine the presence of the 57 kb Northern European deletion in CTNS. Next, the entire coding regions of both DNA strands and the highly conserved exon-intron splice junctions were sequenced. PCR and sequencing were performed at Centogene, The Rare Dissease Company Laboratory, Rostock/Germany. None of the patients carried the 57 kb deletion, but a homozygous missense mutation was found. The reference sequence of the CTNS gene is: NM-001031681.2. A previously reported homozygous mutation c.451A>G (p.R151G) in the CTNS gene was identified in two siblings. The relationship between the detected missense mutation and CTNS was investigated in the Human Gene Mutation Database and the literature.
Two of three Turkish patients with infantile nephropathic cystinosis were genetically evaluated. There was consanguinity in two of the nine families (Figure 1). Cystine crystals were detected in the cornea in all three patients. In cytogenetic analysis, no structural or numerical abnormalities were found in chromosomes in any of the patients. Molecular analysis confirmed that neither of the two brothers carried the 57 kb deletion, but a homozygous missense mutation was found in exon 7 of the CTNS gene (c.451A>G, p.R151G) (Table 1, Figure 3). The mutation found caused an A>G change in the 451st nucleotide of exon 7 (Figure 3). This homozygous mutation was an endemic variant found only in the Mediterranean region of Turkey and specific to the Turkish population [5]. Endemic and non-endemic variants previously found in Turkey are shown in Table 1 and Figure 4. This mutation has previously been described as discribed as disease-causing in Turkish cystinosis patients by Topaloğlu et al. [5,6].
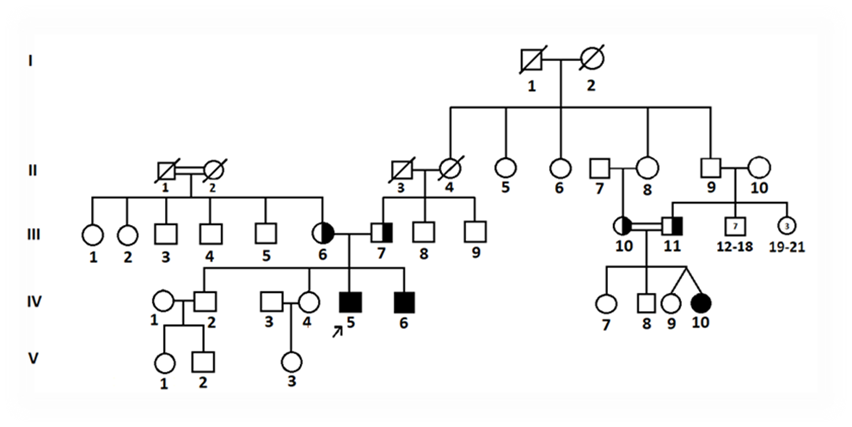
Figure 1. Pedigree of the examined families

Figure 2: Patient general appearance of two brothers after therapy

Figure 3: Schematic representation of the CTNS gene showing the genomic location of the mutation in Turkey. Exon mutations are shown at the bottom of the figure and intron mutations at the top.
| Missense/nonsense mutations | ||||||
| no | mutation | protein | exon | phenotype | country of diagnosis | references |
| 1 | c.15G>A | p.W5* | 3
| Infantile | Turkey Egypt France | Topaloglu et al., 2017, CJASN Kalatzis et al., 2002, Hum Mut Soliman et al., 2014, JIMD Rep |
| 2 | c.451A>G | p.R151G | 7 | Infantile | Turkey | Topaluglu et al., 2012, Pediatr Nephrol Demirhan et al. ** This study |
| 3 | c.470G>A | p.G157D | 8 | Infantile | Turkey | Topaluglu et al., 2012, Pediatr Nephrol |
| 4 | c.518A>G | p.Y173C | 8 | Infantile | Turkey | Topaluglu et al., 2012, Pediatr Nephrol |
| 5 | c.613G>A
| p.D205N
| 9 | Infantile
| Turkey Iran USA | Topaloglu et al.,2017, CJASN Sadeghipour,2017, Hum Genom Var Shotelersuk et al., 1998, Am J Hum Genet |
| 6 | c.664C>T | p.Gln222* | 9 | Infantile | Turkey | Topaloglu et al., 2017, CJASN |
| 7 | c.878G>T | p.S293I 11 | 11 | Infantile | Turkey | Turkey Topaloglu et al., 2012, CJASN |
| 8 | c.922G>C | p.G308R | 11 | Infantile | Turkey | Topaloglu et al., 2012, CJASN |
| 9 | c.1015G>A(•)
| p.G339R*
| 12
| Infantile
| Turkey USA UK Canada Italy Spain Egypt Iran | Topaloglu et al., 2017, CJASN Shotelersuk et al.,1998, Am J Hum Genet. Attard et al., 1999, Hum Mol Genet. Rupar et al., 2001, J Med Genet Mason et al., 2003, Eur J Hum Genet Macias-Vidal, 2009, Clin Genet Soliman et al., 2014, JIMD Rep Sadeghipour, 2017, Hum Genom Var |
| Intronic mutations | ||||||
| no | mutation | intron | phenotype | country of diagnosis | references | |
| 1 | c.141-22A>G | 5 | Infantile | Turkey | Topaloglu et al., 2017, CJASN | |
| 2 | c.140+1G>T | 5
| Infantile
| Turkey UK, France | Topaloglu et alv, 2017, CJASN Town et al., 1998, Nat Genet | |
| 3 | c.681G>A (•) | 10 | Infantile | Turkey Turkey Saudi Arabia Egypt Iran | Döneray et. 2017 Eurasian J Med Topaloglu et al., 2017, CJASN Aldahmesh et al,. 2009, Ophthalm Genet Soliman et al., 2014, JIMD Rep Ghazi et al., 2017, Nephrologia Sadeghipour, 2017, Hum Genom Var | |
| 4 | c.853-1G>A | 11 | Infantile | Turkey | Topaloglu et al., 2012, CJASN | |
| Deletions | ||||||
| no | mutation | protein | exon | phenotype | country of diagnosis | references |
| 1 | c.18_21delGACT (357delGACT)
| T7Ffs*7
| 3 | Infantile | Turkey UK, France Netherlands Italy Mexico France Spain Thailand Iran | Topaloglu et al., 2017, CJASN Town et al., 1998, Nat Genet Heil et al., 2001, Nephron Mason et al., 2003, Eur J Hum Genet Alcantra-Ortigoza, 2008, Hum Genet Servais et al., 2008, CJASN Macias-Vidal, 2009, Clin Genet Yeetong et al., 2012, Gene Shahkarami, 2013, Nephrologia |
| 2 | c.291_294delTACT | T98Ffs*19 | 6 | Infantile | Turkey | Topaloglu et al., 2017, CJASN |
| 3 | c.325_329del | Splicing | 6 | Infantile | Turkey | Doneray, 2017, Eurasian J Med |
| 4 | c.834_842del | 279-281del | 10 | Infantile | Turkey | Topaloglu et al., 2017, CJASN |
| 5 | c. 960del | Y321Tfs*8 | 11 | Infantile | Turkey | Topaloglu et al., 2017, CJASN |
| 6 | 10 kb del, c.62-1083_551 | 4–8 | Infantile | Turkey | Topaluglu et al., 2012, Pediatr Nephro | |
| Insertions | ||||||
| no | mutation | protein | exon | phenotype | country of diagnosis | references |
| 1 | c.829dupA
| T277Nfs*19
| 10
| Infantile
| Turkey Belgium Egypt | Topaloglu et al., 2017, CJASN Besouw et al., 2012, Pediatr nephrol Soliman et al., 2014, JIMD Rep |
*Table is updated up to July 2018 and adapted from citation (10). (mutations cited in the text; * data based on public HGMD database).
Table 1*. Reported mutation types and distribution in the CTNS gene of patients with nephropathic cystinosis in Turkey
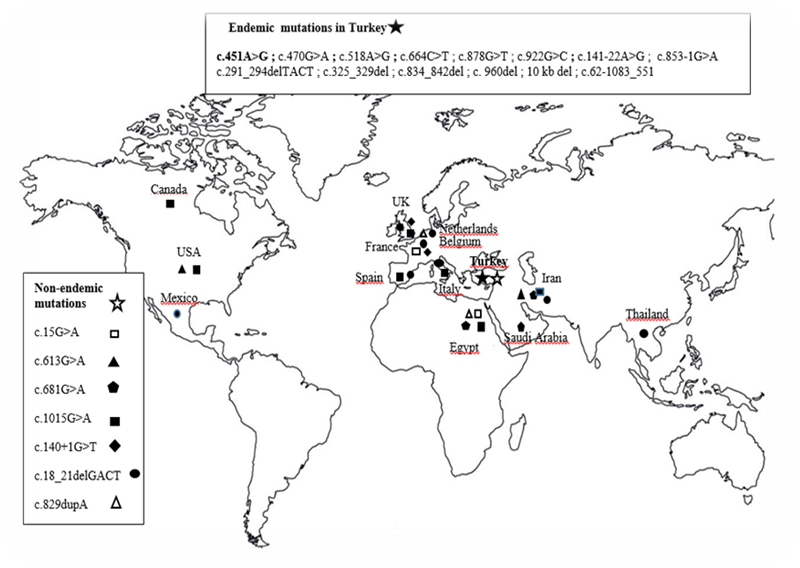
Figure 4: Worldwide geographical distribution of CTNS mutations detected in the Turkish population; Endemics ( ) and non-endemics (
) and non-endemics ( )
)
Elucidating the similarities and differences between patients with cystinosis around the world, early diagnosis of the disease, genetic counseling, prenatal diagnosis and treatment are important in terms of defining the genetic map of the disease in detail. A large number of pathogenic CTNS mutations have been reported in cystinosis patients around the world. However, its incidence and geographical distribution are still not well known. In recent years, an increasing number of reports have been published on CTNS gene mutations in some populations of developing countries. The differences between the genetic background of Turkish, European and North American patients with cystinosis were first reported by Topaloğlu et al. [5,6]. Since the Turkish population is genetically heterogeneous, it can be expected to show different mutations that cause cystinosis. However, some mutations are associated with ethnicity and some are not (Figure 4). The mutations found can cause loss of function of the protein and a severe infantile nephropathic phenotype. To date, approximately 200 different mutations such as missense, nonsense, deletion, insertion and splicesite mutations have been identified in the promoter region in patients with cystinosis.8-18 Most of the reported CTNS mutations have been detected in European and North American countries. In Turkey, there are only two studies on this subject, and endemic-non-endemic variants have been reported [5,6,17]. These variants are shown in Table 1, Figure 4 and Figure 5; 13 of them are endemic and 7 are non-endemic. But the birth prevalence of cystinosis in Turkey is unknown, it has been reported that the general incidence rates in the populations of France, Australia, Germany, Denmark and Sweden are 1:167.000, 1:192.000, 1:179.000, 1:115.000 and 1:260.000, respectively [16,18-24]. To date, the highest birth frequency has been reported in the Pakistani ethnic group living in the West Midlands region of the United Kingdom (1:3,600) [24]. Since cystinosis is an autosomal recessive disease, its incidence is affected by the degree of consanguinity in the society. Accurate statistical date on the incidence of cystinosis is lacking in regions with high levels of consanguineous marriage, such as the Middle East and North Africa. However, different lysosomal storage disorders were reported to be found in 13.7% of a large patient cohort in Egypt [25,26]. However, the genetic spectrum of cystinosis patients in large geographical regions such as Sub-Saharan Africa, Southeast Asia and the Far East is not well known. Studies conducted in the Middle East, Mexico and South Africa have reported that the incidence of cystinosis is higher in most of these countries than in Europe and North America [13, 19,27-31]. In poor countries, many cystinosis go undiagnosed and patients die young due to complications of the disease. We estimate that the disease will be much higher in our country due to the high rate of consanguinity. This report is considered the third study on the molecular analysis of CTSS, after the two studies by Topaloğlu et al. [5,6] and Döneray et al. [17]. Molecular analysis of the CTNS gene in our cases confirmed the diagnosis of the disease. Previously, a study by Topaloğlu et al revealed that the Turkish cystinosis population had new genetic features. The most frequently detected pathogenic mutation is a 57 kb deletion present in patients of Northern European and North American origin. However, the mutation is almost completely absent in this geographical distribution, especially in the Middle East, including Turkey [8,5,12]. In the present study, we could not find the 57 kb deletion in the CTNS gene in the Mediterranean region of Turkey. However, the mutation spectrum of CTNS appears to be different in Mediterranean and Middle Eastern populations. Meanwhile, small deletions and point mutations were seen in a population originating from Saudi Arabia.[12]. Many different mutations throughout the gene have been reported in European and American cystinosis patients [2,10]. The most common was the large 57 kb deletion [31-33]. This deletion was detected at a very low rate in Italians, French Canadians and Mexicans [23,33]. This carrier rate was 17% in Italians. It was reported that this large deletion was not found in any of the Iranian and Egyptian patients [28,22]. As a matter of fact, the 57-kb deletion has been done before in the Turkish population and has not been reported from the world's largest pediatric cystinosis registry [5,6]. The absence of the 57 kb deletion in our patients and in previous studies conducted in Turkey supports that the Turkish population did not mix with the European gene pool. Numerous studies have been conducted on the role of CTNS mutations in various forms of cystinosis. Many studies have been conducted on the genetic basis of cystinosis with European and American patients with cystinosis, and many mutations have been identified. Serious CTNS mutations, including deletions, insertions, nonsense, missense, and splicing mutations, have been reported in patients with infantile nephropathic cystinosis. More than 200 pathogenic CTNS mutations have been reported in cystinosis patients worldwide. There is a genotype-phenotype correlation associated with clinical forms of cystinosis. Severe mutations in both alleles are usually associated with the infantile severe form of the disease. Juvenile and ocular forms of cystinosis are usually associated with a mild mutation. Some mutations also lead to residual function of cystinosin in intermediate or adult patients. Missense mutations are often found in individuals with less cystine accumulation in their leukocytes. A mild mutation is found in patients with moderate cystinosis (i.e., nephropathic but late onset) [35]. Of the mutations seen in Turkey, 13 are endemic and 7 are endemic, except for the 57 kb deletion (Figure 3) [5,6]. In one of the largest studies conducted in Turkey, it was reported that no patient had the 57 kb deletion and that the deletion was limited to the Eastern Mediterranean and the Middle East [5,6]. In another study, homozygous c.681G>A (p.E227E) and homozygous [c.325_329del (p.Thr109ProfsX14)] mutations compatible for a 5 bp deletion were reported in two Turkish patients [6] (Table 1). In this study, a previously reported homozygous mutation (c.451A>G p.R151G) in exon 7 was detected in the brothers. The parents of these children were also compound heterozygous for the missense variant c.451A>G (p.R151G).This missense mutation was first described by Topaloğlu et al. [5,6] in the Mediterranean region of Turkey. Frameshift and nonsense mutations lead to protein truncation and were predicted to be easily pathogenic. It was determined that c.451A>G in patients was pathogenic when arginine was replaced by glycine at position 151, which is a highly conserved residue among different species. It is predicted that this mutation may result in a structurally defective CTNS protein. This mutation (c.451A>G (p.R151G) was not detected in the Saudi, Iranian and Egyptian population. This is quite remarkable, considering the long-standing common relations between these three close/neighboring countries. The found c.451A>G sequence variation may disrupt the consensus sequence and interfere with correct or efficient splicing. One of the two families with a sick child was the child of an aunt and uncle. A study including 86 adult patients with cystinosis reported a lower parental consanguinity rate (14%) and a mean age of diagnosis of 1.5 years [6]. They also reported that the earliest clinical symptoms of cystinosis are polyuria-polydipsia, followed by growth failure and rickets. As a matter of fact, the most common clinical findings in our patients were polyuria, polydipsia and growth retardation. Similarly, rickets was observed at the time of diagnosis in almost half of the patients in this study. Both siblings homozygous for the missense variant c.451A>G (p.R151G) had an intermediate disease course, reaching puberty with proteinuria but no renal failure. The patient without molecular analysis had similar clinical findings. This mutation in the CTNS gene may lead to loss of function of the protein and cause the infantile nephropathic phenotype as observed in patients. The CTNS mutation was important to our patient's family because they were planning a fifth pregnancy. The parents also carried the same mutation heterozygously, and the family's chance of having a child with nephropathic cystinosis was around 25% for each pregnancy. Since consanguineous marriages are high in the Turkish population, the rate of mutations causing nephropathic cystinosis may be high. The most common form of cystinosis, the infantile type, manifests itself by the age of 10 with renal failure, other systemic complications, and early corneal cystine crystal deposition. Deposition of corneal crystals is common and manifests itself in patients with photophobia, which is a major ocular symptom [36]. Cystine crystals also affect the conjunctiva and retinal pigment epithelium. It can also accumulate in the iris, ciliary body, choroid and lens capsule. The most common clinical findings in the current study were growth failure, polyuria, polydipsia, vomiting, loss of appetite, rickets, eye retention, and renal failure. In summary, our results once again confirm the broad mutation spectrum of the CTNS gene, showing that the 57 kb deletion is not seen in the Mediterranean region and is not found in the Turkish population. At the same time, our findings support the relationship between CTNS mutations and cystinosis, and will form the basis for further research in the Turkish population. We show that Turkish cystinosis patients have endemic CTNS variants compared to patients from European, North American and Middle Eastern countries. It shows that the mutation identified in our case is a previously identified mutation and can be considered a specific mutation specific to the Turkish population. Systematic collection of CTNS mutations in cystinosis patients will aid in the characterization of the disease spectrum worldwide and facilitate comparison of patients from different geographical regions. The mutation spectrum in Turkish cystinosis patients should be investigated in future studies.
All authors have no conflict of interest.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
