
case report | DOI: https://doi.org/10.31579/2692-9406/125
¹Prison Health Center, ”Mother Theresa University Hospital” Tirana Albania.
²Prison Health Center, “Mother Theresa University Hospital” Tirana, Albania.
*Corresponding Author: Marsela Shani, Prison Health Center ,”Mother Theresa University Hospital” Tirana Albania
Citation: Marsela Shani and Drita Jaka (2022) Morbus Gaucher a diagnostic challenge. J. Biomedical Research and Clinical Reviews, 7(3); DOI: 10.31579/2692-9406/125.
Copyright: © 2022 Marsela Shani, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 05 July 2022 | Accepted: 23 August 2022 | Published: 05 September 2022
Keywords: morbus gaucher; β-glucocerebrosidase; enzymatic activity; splenomegaly; thrombocytopenia; ferritin; transferrin saturation
Morbus Gaucher is an inherited disease of fat deposition caused by an autosomal recessive defect in the gene encoding the enzyme β-glucocerebrosidase, responsible for the accumulation of glucosylceramides in reticuloendothelial cells, turning this multidimensional disease into a debilitating Gaucher disease and are found mainly in the spleen, liver, bone marrow and rarely in the lung. The level of glucocerebrosidase enzymatic activity in patients with Gaucher is seen to reach around 5–25% of normal activity. Measurement of this level in leukocytes or cutaneous fibroblasts is considered the “Gold Standard” for Gaucher diagnosis. Although Gaucher is the most common lysosomal deposition disease it remains rare and most cases present with a gradual installation of the clinic which explains the delay in diagnosis. It is very important to include Gaucher as a possible diagnosis in cases of splenomegaly and / or thrombocytopenia. The study, "Predicting the probability of Gaucher disease in subjects with splenomegaly and thrombocytopenia," published in Scientific Reports, managed to reconstruct a diagnostic algorithm that managed to identify the relationship between platelet count, ferritin and transferrin saturation, thus making it possible to achieve a possible approach regarding the diagnosis of GD, which for the sake of truth continues to be an undiagnosed / misdiagnosed pathology.
Morbus Gaucher is an inherited disease of fat deposition caused by an autosomal recessive defect in the gene encoding the enzyme β-glucocerebrosidase, responsible for the accumulation of glucosylceramides in reticuloendothelial cells, turning this multidimensional disease into a debilitating Gaucher disease and are found mainly in the spleen, liver, bone marrow and rarely in the lung. The responsible find is on chromosome 1q21 and contains 11 exons and 10 introns, with about 7.6 kilobases (kb).
GD is pan-ethnic, but has a higher prevalence in Ashkenazi Jews. In this group non-neuropathic forms have a frequency of about 1: 850 and the carrier frequency reaches 1:17. Acute neurological forms are even rarer. The prevalence of 1: 500000 is considered low although fatal neonatal cases (fetal hydrops, congenital ichthyosis) fail to be diagnosed.
There are three main clinical forms:
Type I - or adult form characterized by anemia, thrombocytopenia, splenomegaly, skeletal disorders, and in a small number of patients is encountered pulmonary involvement and pulmonary hypertension. Clinical aggression varies from mild to moderate, while the activity of glucose is moderate reduced
Type II or infantile form occurs in 1 in 100000 live births. The clinic is dominated by nervous system problems associated with exitus at an early age (before the age of 2 years) with almost minimal glucocerebrosidase activity
Type III or otherwise called juvenile, occurs in 1 in 50,000 live births, with late installation of neurological problems dominated by incoordination, myoclonic crisis, with slow progression which becomes sever in late childhood. Small enzymatic activity.
Diagnosis: The level of glucocerebrosidase enzymatic activity in patients with Gaucher is seen to reach around 5–25% of normal activity. Measurement of this level in leukocytes or cutaneous fibroblasts is considered the “Gold Standard” for Gaucher diagnosis.
Beta-glucosidase activity
Beta-glucosidase activity was studied in Ospedale Gaslini, Genoa, Italy. Normal values range from 4.4 to 17.7 pmol / punch / h. Subjects exhibiting activity below 4.4 pmol / punch / h were retested for enzyme activity in leukocytes, EBV lymphoblasts, or fibroblasts.
As with other lysosomal enzymopathies, reduced values of the measured enzymatic activity lead to the need to identify the relevant genetic defect. A 5.7-kilobase Gaucher pseudogen with the same number of exons and introns of about 16 kilobases similar has been identified with the Gaucher gene in about 96% of it in all coding regions. To date, about 300 mutations of the Gaucher gene have been identified, most of which are point mutations. There are four mutations that are thought to play a special role in the phenotype of pathology, N370S, 84GG, L444P and IVS2 + 1G. Genetic diagnosis can be made during pregnancy using amniocentesis or chorionic villi but this is only known to the population with high risk for Gaucher.
Biopsy
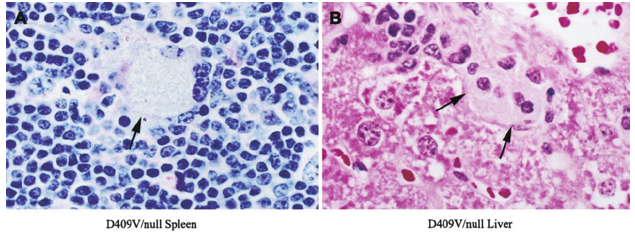
Diagnosis of Gaucher in biopsy materials of different tissues is not a diagnostic criterion. In fact, about 60% of all cases are still diagnosed by bone marrow biopsy or spleen histology after surgical procedures.


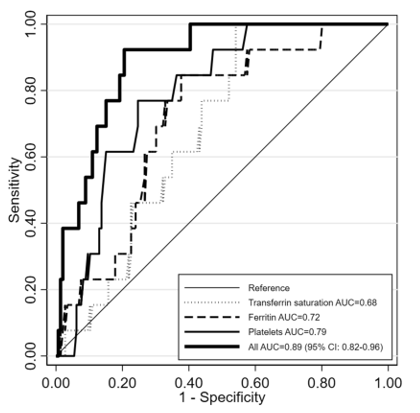
As lysosomal deposition diseases continue to be misdiagnosed or diagnosed late, this brought to the attention of researchers the creation of possible correlations between diagnostic elements that could speak in favor of GD, building genuine mathematical models which could create clues about a possible Gaucher diagnosis.Eventually newborn screening has been experimented in several countries with different results according to the ethnicity of the test population. The study, "Predicting the probability of Gaucher disease in subjects with splenomegaly and thrombocytopenia," published in Scientific Reports, managed to reconstruct a diagnostic algorithm that managed to identify the relationship between platelet count, ferritin and transferrin saturation, thus making it possible to achieve a possible approach regarding the diagnosis of GD, which for the sake of truth continues to be an undiagnosed / misdiagnosed pathology.
Among the enrolled subjects, the only clearly different parameters between GD1 and non-GD1 patients were serum platelet count and ferritin. Hyperferritinemia with normal transferrin saturation is a common finding in de nuovo patients with GD120, with a prevalence ranging between 63 and 81%. Finally, a new diagnostic diagram has been proposed, which enhances the role of hyperferritinemia when associated with splenomegaly and thrombocytopenia.
Scientific Reports (Sci Rep) ISSN 2045-2322 (online)
As can be seen from the diagram high levels of ferritin coupled with normal transferrin saturations are a diagnostic element with high sensitivity in GD,platelets as well also.
Differential diagnosis is mainly made with other deposition diseases or with diseases of the hematological spectrum. Although pseudo-Gaucher cells are not distinguished from true cells by hematoxylin-eosin staining, iron staining is generally negative in pseudo-Gaucher.
Niemann-Pick is a sphingomyelinase deficiency disease, which leads to the accumulation of sphingomyelins in the macrophage cytoplasm. The cells are small and usually uniform in size. The cytoplasm is foamy and vacuolated as in Gaucher cells.
In Tay-Sachs (hemoaminidase A deficiency), there is an accumulation of GM2 in the heart, spleen, and liver. vacuolated is predominant.
Pompe disease (maltase deficiency) has glycogen deposition in hepatocytes and muscle cells but the primary damage is to the skeleton and heart muscle without involvement of the bone marrow.
Blue histiocytosis is associated with massive intramedullary destruction as in the LMC. The macrophage cytoplasm is filled with insoluble fatty pigments called ceroid.


A case: F.B 57 years old, diagnosed with diabetes for 3 years, under treatment with oral diabetes. The only complaint of fatigue accompanied by bone pain for almost 8 years. Reports that he has had bone pain all his life and has often received treatment with cortisone. being classified as generalized arthritis. Examinations show:
Objective examination
C-V system: FC = 88 / min; TA 110/70 mmHg, no pathological noise, normal ECG
Vital signs: Temp. 36.3 C, Sat O2 98% (RA)
Respiratory System: Normal broncho-vesicular respiration
On palpation: Treatable soft abdomen, liver 1 cm below the costal arch, spleen 3-4 cm below the costal arch ,genitourinary tract normal.Normal reflexes present without pathological findings.
Blood count: Rbc: 4.100.000 / mm3 Hgb: 7.8 g / dl Hct: 22.5% PLt: 87,000 / mm3 Wbc: 6.300 / mm3 lymphocite = 34% monocite = 4% granulocite = 52%
Biochemical values : Glucose: 172 mg / dl, Urea: 45.5mg / dl, Creat: 0.67mg / dl, AST: 23U / L, ALT: 29 U / L, Bil.tot: 1.1 mg / dl, Prot tot: 6.7 g / dl
Abdominal ultrasound: Liver is normoechogenic, d.max: 138mm, gallstone with no stones, free bile ducts, the pancreas is normal, normal kidneys, spleen with d.max-165mm,urinary tract and normal prostate. Minimal liquid is found around , spleen and in Douglas area.
HIV Ab –neg; HBsAg neg, HBcAb-neg; HBcAb-neg; HCVAb neg; Wright test - neg; LDH = 512; Ferritin = 5677; Trasferine saturation 427 mcg/dl
Bone Marrow biopsy: Focal clusters / diffuse replacements with oval histiocytes 25-80 microns with abundant pale gray, blue cytoplasm with multiple folds. Small nucleus with dense chromatin, marked increase of reticulin fibers. Small nucleus with thick chromatin and fuzzy nucleus. Histiocytic cell content with abundant cytoplasm. The presence of vacuoles is observed. The appearance suggests an accumulating lysosomal disease matching, the Gaucher cells.

Although Gaucher is the most common lysosomal deposition disease it remains rare and most cases present with a gradual installation of the clinic which explains the delay in diagnosis. It is very important to include Gaucher as a possible diagnosis in cases of splenomegaly and / or thrombocytopenia. GD is characterized by its clinical polymorphism.
In correlation with neurological involvement and severity of the disease are classified 3 main forms of the disease.
Type I non-neuropathic form is found mainly in adults
Type II neuropathic form acute form encountered mainly in newborns with almost 100% mortality
Type III neuropathic form mainly subacute or juvenile form
GD type I is the most reported clinical form although Gaucher is considered a rare pathology. Also GD type I refers to have the best survival rate of GD and the onset of symptoms may be at an early age, often with symptoms that attributed to a myriad of other diagnoses. In general, the main clinical manifestations include severity in the left hypochondrium due to considerable splenomegaly and skeletal-related symptoms. Association with hepatomegaly often leads to significant abdominal distension, anemia and thrombocytopenia in the field of splenic sequestration, which clinically translate into fatigue, weakness, epistaxis or even the presence of cutaneous hemorrhagic phenomena such as hematoma, ecchymosis, petechiae, etc. Gaucher cell marrow identification although the Gold Standard for diagnosis is the measurement of the level of enzymatic activity.
Differential diagnosis should be made with diseases such as Fabry disease, Gangliosidosis, Wolman / cholesterol ester, Niemanne Pick A and B (dark blue histiocytes) and hematologic disorders with pseudo-Gaucher cells such as: multiple myeloma, Hodgkin's lymphoma, Hodgkin's lymphoma jo-Hodghoma. myeloid leukemia, myelodysplasia, acute B-cell lymphoblastic leukemia, thalassemia and sickle cell disease.
More than 300 different mutations have already been identified from the Human Gene Mutation Database. The most common is the substitution in the N370S allele. Subsequent substitutions at L444P and 84 GG. Patients may be affected by 2 or more mutations at the same time classifying these cases as composite heterozygosity. These mutations have been found to be present in about 70% of European patients.
The L444P mutation occurs as a result of the replacement of the thymus fibrosin in nucleotide 6433 of the active glucose cerebrosidase gene, creating a new space for the restriction enzyme Nci I, the replacement of proline by leucine. This is the most common mutation in patients with neurological symptoms of Gaucher disease.
Enzyme-driven macrophage replacement therapy (TRE) has long been the standard treatment. We are not talking here about a treatment which finally solves the Gaucher problem, as at the basis of this disease lies a genetic defect.
From the above it turns out that Gaucher is an undiagnosed or little diagnosed disease. Therefore such a situation requires increased attention of clinicians related to Gaucher considering it a potential diagnosis especially in hepatosplenomegaly associated with anemia / thrombocytopenia there are other causes which would justify the clinic. Equally delicate is the situation when we talk about differential diagnosis of Gaucher due to a multitude of pathologies with histopathological similarities to it.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
