
Review Article | DOI: https://doi.org/10.31579/2642-973X/135
1 -Dental Surgeon (DDSc),
-Oncologist (MSc), Specialized in Clinical Oncology, Cytology and
Histopathology, Dept. of Pathological Anatomy, Medical School,
University of Athens, Athens, Greece
-Resident in Maxillofacial and Oral Surgery, 401 General Military
Hospital of Athens, Athens, Greece
-PhD in Oncology (cand)
-Registrar in Dentistry, NHS of Greece
2 -Colonel – Neurosurgeon (MD),
-Director of Neurosurgery Dept., NIMTS Military Hospital of Athens, Greece.
*Corresponding Author: Nikolaos Andreas Chrysanthakopoulos, Dental Surgeon (DDSc), -Oncologist (MSc), Specialized in Clinical Oncology, Cytology and Histopathology, Dept. of Pathological Anatomy, Medical School, University of Athens, Athens, Greece.
Citation: Nikolaos A. Chrysanthakopoulos, Panagiotis A. Chrysanthakopoulos, (2025), Molecular Biology of Gliosarcoma-An Essential Review, J. Brain and Neurological Disorders, 8(1): DOI:10.31579/2642-973X/135
Copyright: © 2025, Nikolaos Andreas Chrysanthakopoulos. This is an open-access article distributed under the terms of The Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 13 February 2025 | Accepted: 21 March 2025 | Published: 28 March 2025
Keywords: gliosarcoma; glioblastoma; molecular biology; mutation; genetics
Gliosarcoma (GSM) is a very rare brain neoplasm, a histologic variant (IDH-wild type phenotype of Glioblastoma Multiforme (GBM), and like GBM is characterized by a poor prognosis compared to other Grade IV gliomas. The median survival of GSM is less than one year, whereas less than 5% of GSM patients survive after 5 years after performing the conventional therapy such as surgery, chemotherapy and radio treatment. Although it has similarities to GBM, GSM displays diverse distinct differences, morphologically and molecularly. It is a highly aggressive primary brain tumor with histologic components which comprise of glial (astrocytic) and sarcomatous features. Several differences have been observed in histological and molecular elements, however, detailed data regarding the genetic background of GSM is lacking. Most of GSMs are sporadic, however it is irrefutable that a minor percentage has been associated with germline mutations and various inherited cancer susceptibility syndromes, such as Lynch Syndrome. Previous reviews have demonstrated that GSM carries somatic changes in genes coding for PI3K/Akt (PTEN, PI3K) and RAS/MAPK (NF1, BRAF) signaling pathways that are critical for tumor development. It is important to notice that the PTEN alterations frequency in GSMs was greater than in GBMs. Various novel translocations, such those in the RABGEF1 gene, which create probably adverse combinations have been observed.19 conventional genes have been detected in GSM, determined as those changed in more than 5% of samples, including PTEN(66%), TERT promoter (92%), and TP53 (60%). EGFR and CDKN2A also exhibited alterations in GSM cases. Tumors with available molecular profiling were mainly MGMT-un-methylated (87.5%), EGFR wild-type (100%), and IDH-1-preserved (100%). The current review highlights important molecular biology features of GSM in the light of recent literature, including its histological characteristics.
GSM is a primary malignant brain tumor which shows high heterogeneity, invasiveness, and resistance to modern treatments. It is considered to be a distinct clinicopathological disease [1,2] in the central nervous system (CNS) tumors classification and comprises approximately 2% of all the glial malignant neoplasms [2-4], represents less than 0.5% of all intracranial tumors and is most common in adults between 40 and 60 years old. GSM incidence has been estimated between 1% and 8% of all malignant gliomas, constituting only 0.48% of all brain tumors and from 1.8% to 2.8% of GBM cases [5-9], and with a low incidence of 0.59%-0.76% among all adult brain tumors [10]. Males are affected more frequently, than females (M:F ratio 1.8:1) [1,5, 11]. In pediatric individuals, it is infrequent, whereas it is more common in the white and non-Hispanic population [6, 9,12, 13].
GSM is regarded as grade IV neoplasm and is classified as a GBM variant in the revised 2007 WHO classification [14-16]. GSM was first mentioned by Heinrich Strobe in 1895 as a brain tumor comprising of both glial and mesenchymal ingredients [17].
GSMs are further categorized into primary de novo and secondary GSM, which are characterized by different median survivals between both types (25 vs. 53 weeks) [6]. Secondary GSM is believed to have appeared as a recurrence or progression of GBM, or as a consequence of radiation therapy [6,12]. Primary GSMs commonly appear de novo with a preference for the temporal lobes, whereas secondary GSM occur after cranial radiation for GBM, as mentioned [3,12]. The dura invasion and extracranial metastases were more frequent in GSM than GBM with possible prognostic consequences [3,18], although some reports detected no considerable differences regarding the overall survival (OS) between both diseases [6, 19]. GSM mainly affects supratentorial locations and is localized in the temporal and parietal lobes, followed by the frontal and occipital lobes [14,20]. GSMs affecting the spinal cord are rare, constituting about 1% of all malignant spine gliomas [21] and may indicate metastasis comes from intracranial tumors or less commonly, de novo development [22, 23].
In clinical level, GSM progresses quickly and patients show a 3% greater risk mortality as compared with GBMs [24]. Prognosis of GSM is similar to GBM with a greater extraxial metastases incidence being detected [25]. Cachia et al. observed that the primary GSM median OS was 17.5 months [12]. Another multi-center report assessed the GSM median OS as only 13 or 15 months, and also stated that chemotherapy with temozolomide (TMZ) was not resulted in an improvement in OS compared to radiation [26]. Similar research showed that even after standardized therapy, the mean OS time was only 6.6-18.5 months [27]. GSM is also characterized by a great rate of recurrence and metastasis. Although GSM management is in general similar to GBM, several clinical perspectives such as a tendency to extra-cranial metastasis, distinct radio-logical features and worse prognosis than GBM, indicate that GSM may be a distinctive clinicopathological disease [6].
Malignant astrocytes represent the majority of the glial element in GSMs, however, oligodendroglial elements have also been reported. GSM is a CNS mixed primary neoplasm, constituted of astrocytic anaplastic and malignant mesenchymal components [6,18, 28-32]. The gliomatous component exhibits GBM elements as it is anaplastic, often spatially distinguished, characterized by the dura and leptomeninges invasion, and hyperplastic or hypertrophied blood vessels. The gliomatous component also expresses glial fibrillary acidic protein (GFAP) and is reticulin-poor, whereas sarcomatous component is reticulin-rich but does not express GFAP [12]. The sarcomatous component shows malignant transformation signs such as mitotic activity, atypia of nucleus, and bundles of spindle cells. In some GSM cases mesenchymal differentiation with collagen deposition have been revealed [33].
According to histology, the glial element accomplishes the GBM cytologic criteria, and the mesenchymal element may exhibit a large diversity of morphologies with origin from fibroblastic, osseous, cartilaginous, striated and smooth muscle, or adipose cell origin. Conventionally, sarcomatous components resemble fibrosarcoma or malignant fibrous histiocytoma. The mentioned biphasic tumor subsequently was accepted as a result of the detailed histological analyses by Feigin et al. [34].
Because of the lack of particular and consistent diagnostic criteria however, the term GSM was also concerned tumors of glial origin which have acquired mesenchymal phenotypes, such as the ability to produce collagen fiber and reticulin network [34]. As mentioned, those tumors consist distinct diseases, the first as a glial origin tumor with mesenchymal components, known as glioma with desmoplastic metaplasia or desmoplastic glioma, and the second as a tumor with distinct gliomatous and sarcomatous components, known as GSM [35]. Other types of rare GSM transformation’s concern osteosarcoma, angiosarcoma, chondrosarcoma, and liposarcomatous, leiomyomatous, myosarcomatous, and neuroectodermal tumors [32,36-40].
The mesenchymal elements would be diagnosed as fibrosarcoma or undifferentiated pleomorphic sarcoma in a soft tissue microenvironment, whereas chondroosteogenic and myogenic differentiation may also be found in that microenvironment. Variants which contain liposarcomatous, angiosarcomatous, and mixed mesodermal-type characteristics have also been observed. Squamous differentiation, grandular structures, and adenoid development may also be exhibited within the glial locations of selected cases [11].
The accurate GSM etiopathology remains unknown. It has been suggested that the sarcomatous component arises from the hyperplasic blood vessels malignant transformation, frequently found in high-grade gliomas [41]. Brain neoplasms, comprising GSMs, are mainly sporadic, and only a small rate of those have been associated with hereditary cancer susceptibility syndromes, such as Lynch Syndrome (LS) [42]. LS consists an autosomal dominant tumor syndrome with a prevalence of about 3-5% of all bowel cancers. LS is also able to increase the developing tumors risk in the colorectum and other organs, such as the gastrointestinal tract, liver, gallbladder, ovaries, endometrium, brain, upper urethra, skin, etc. [43]. It has been recorded that the primary brain tumors risk, especially high-grade gliomas, increases by about four times in LS patients. However, few clinical cases have confirmed the relationship between LS patients and GSM appearance [42]. The majority of those neoplasms appear without the presence of known predisposing factors, however they have also been associated with prior irradiation, including the Thorotrast intra-cranial instillation [11]. The essential signaling pathways involved in GSM pathogenesis are presented in Figure 1.
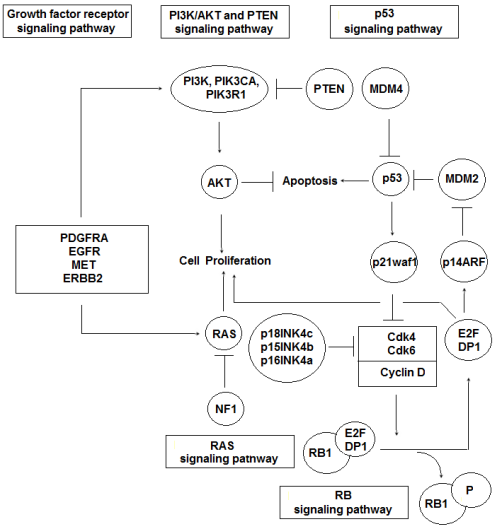
Figure 1: Essential pathways involved in GSM pathogenesis
*Displays truncating mutation caused by early stop codon
The GSM contemporary treatment is similar to the treatment used for GBM and is mainly surgical, combined with postoperative chemotherapy and radiotherapy, however, the clinical out-comes remain poor with 5-year survival rates below 10% [44], and a median survival of 9 months compared with a median 15-month survival for other GBM forms [5].
Molecular Biology
The majority of brain tumors, including GSM, is in most cases non-hereditary and is mainly influenced by somatic gene mutations and various environmental factors. Frequent mutations have been detected in TP53, TERT, PTEN, and NF1genes in GSM cases, which have been linked with known cellular functions such as cell cycle regulation, genetic stability, and cellular proliferation [45]. Similarly, in a genetic analysis by Zaki et al. [9] was detected that the most commonly genes with mutations in GSM were TP53 (60%), PTEN (66%), TERT promoters (92%), and NF1 (41%), indicating the complicated and various GSM pathogenesis.
GSMs and primary GBMs have similarities in their molecular profiles and show a similar ratio of PTEN, RB1 and NF1, changes. However, TP53 mutations are more common and the ratio of EGFR overexpression/amplification is lower in GSM as compared with GBM [12, 18,46]. Recent reports have examined the genetic changes in primary and secondary GSM and molecular analyses revealed a great TP53 mutations incidence and, scarcely, IDH and EGFR mutations [4,12,18,47,48], whereas similar researches reported lower frequencies of TP53 mutations [49,50]. GSM has been diagnostically separated into TP53 mutated and wild type GSM variants [18]. In GSM diverse unique copy number alterations have been detected and a subsection of changes presented especially in the sarcomatous element. Genetically, GSM is unstable, with a high rate of heterozygosity loss at 10q (88%) [31]. It has also been found that the GSM monoclonal origin would be linked with the p53 mutation, recorded in 23% of GSM compared with 11% of primary GBM, and showed also the p16 deletion [9,49,51].
Recently, few studies of concise genome sequencing of GSMs, that have mentioned key somatic mutations in known oncogenes such as TP53, RB1, PTEN, and NF1 and also amplifications of EGFR, AKT1, PDGFRA, MDM2, CDK4/6, and MET genes have been carried out [18,52,53]. RB1 gene somatic alterations have been observed in 30% of GSM samples. (Table 1).
Mutations have also been observed in great frequency in GSM specific analysis concerned TERT promoter, STAG2, and CDK2NB. Overall, the mentioned mutations are characterized by unclear clinical and prognostic coherence, though represent an interesting pathway for further growth as prognostic or tumor-specific treatment markers. Previous reports have found that the RAS/ MAPK and PI3K/Akt pathways alterations are critical for GSM development [54].
PTEN alterations have been recorded in 26% of high-grade gliomas in the TCGA data, and identified in 45% of examined GSMs cases [52,55]. The incidence of TERT promoter mutations in GSM has been estimated to be 83% [31], mutation of PTEN varies from 28.6% to 45% and homozygous deletions of P16 INK4alpha was 37% [38,40]. EGFR amplification was observed in 4% of GSM cases [40], whereas IDH mutations were rare [38].
Cho et al. compared GBM with GSM using whole exome sequencing and copy number variants (CNV), and observed that the following pathways more frequently changed in GSM, such as TP 53, PTEN, EGFR, RAS/MAPK, PI3K/Akt, RASGRF2, PAK3, ITGB7, FGFR1, COL5A1. The authors also found more frequent changes in phosphatidylinositol/calcium signaling (CACNA1F/ 1I, PLCB3/L1, ITPR1/3) [18].
From a bio-molecular point of view, GSM carries mutations in common with sarcoma of soft tissue caused by complication in the TP53, TERT promoter, Cyclin-dependent kinase inhibitor 2A(CDKN2A), CDKN2B, Retinoblastoma associated Protein Type 1(RB1), and Neurofibromin 1 (NF1) [49]. Similar to GBM, GSM inhibits mutations in EGFR, PTEN, STAG2, and Protein Tyrosine Phosphatase Non-Receptor Type 11 (PTPN11) [5,7,13]. (Table 2)
In 2000, Reis et al. carried out a GSM detailed genetic analysis, searching for mutations which were frequently found in GBM. Using polymerase chain reaction (PCR) single-strand conformation polymorphism analysis and direct sequencing, TP53 and PTEN genes were analyzed, whereas the EGFR, p16, CDK4, and MDM2 genes were examined via differential PCR. With the exception of absence in amplification in EGFR, GSMs exhibited all of the genetic aberrations observed in primary GBM (TP53 and PTEN mutations, MDM2 and CDK4 amplification, homozygous p16 deletion). In addition, identical mutations in TP53, p16, PTEN, and CDK4 genes were recognized in the glial and sarcomatous elements of a GSMs subset examined [46]. (Table 1). Most GSMs carry somatic changes of RAS/MAPK (NF1) and PIK3/Akt (PTEN, PI3K) signaling pathways which are critical for tumor development [54]. PTEN somatic mutations/ indels were revealed in 50% of specimens and this frequency was greater than previously recorded,14% and 38%, respectively [12,46].
| Tumor | Genetic Alterations* |
| Soft Tissue Sarcoma (STS) | TERT, DOCK8 |
| Glioblastoma Multiforme (GBM) | PDGFRA, PIK3CA, ARID5B, IDH1, PIK3R1+13 mores |
| Gliosarcoma (GSM) | BRAF, SOX2, FBXW7, MSH6, SUZ12+4more |
| STS and GBM | CDK4, ATRX, COL7A1, GLI1, MDM2, KMT2D |
| GSM and GBM | PTEN, EGFR, STAG2, PTPN11 |
| STS and GBM and GSM | TERTp, CDKN2A, CDKN2B, TP53, NF1, RB1, |
Table 1: Usually altered genes in Glioblastoma (GBM), Gliosarcoma (GSM), and Soft Tissue Sarcoma
*Mei-Yee Kiang K, Chan AA, Ka-Kit Leung Gilberto. Secondary gliosarcoma: the clinic-pathological features and the development of a patient-derived xenograft model of gliosarcoma Kiang et al. BMC Cancer (2021) 21:265. https://doi.org/10.1186/s12885-021-08008-y
Actually, together with focal deletions, the somatic alterations frequency in PTEN gene was found 70%, indicating that the alteration of PTEN is essential for GSM growth. PTEN is a mitogenic signaling mediated by class 1phosphatidylinositol 3-OH kinase (PI3K) negative regulator. PTEN gene mutations or deletions occur often and have been linked with therapeutic resistance in GSM [13]. PTEN gene is also mutated/deleted in mesenchymal GBMs in approximately 50% of cases as reported by the TCG [13], whereas PTEN was the most frequently altered gene in GSM cases, as 70% of specimens carried somatic mutations, indels or focal deletions in the PTEN gene location [56,57].
Whereas the pathogenesis of GSM remains poorly understood, several reports have revealed shared mutations and cytogenetic aberrations, such as mutationsof p53 and PTEN, deletion of p16, amplifications of MDM2 and CDK4, between gliomatous and sarcomatous components of discrete tumors, suggesting a monoclonal origin implicating improper gliomagenic cells mesenchymal differentiation [46,52,58,59]. Although it has been subsequently demonstrated that p53 protein immuno-histochemical accumulation does not definitely regard as identical to mutation of TP53 gene, Frandsen et al. initially suggested the possibility of similar TP53 mutations in the two components of GSM based on the p53 immuno-histochemical accumulation in their GSM cases [4]. Eventually, Biernat et al. showed identical TP53 mutations in the glial and sarcomatous elements of two GSM cases via single-strand conformational analysis and direct DNA sequencing [59].
The GSM sarcomatous component histogenesis has been found to be controversial. Previous studies indicated that the sarcomatous elements derived from the hyperplastic blood vessels neo-plastic transformation, commonly observed in high‑grade gliomas. Genetic researches detected the identical p53 and PTEN mutations presence and similar chromosomal abnormalities and cytogenetic changes in GSM glial and sarcomatous elements components indicating a monoclonal origin [46]. Paulus et al. using interphase cytogenetics, i.e., in situ hybridisation, exhibited monosomy for chromosomes 10 and 17 in both the glial and sarcomatous components of GSM, suggesting also a monoclonal origin for both components [58].
Minor differences between GBM and GSM in PTEN mutations and CDK amplification were observed in glial and sarcomatous components [60]. Moreover, less than 12% of GSMs have O6-methylguanine-DNA methyl-transferase gene promoter (pMGMT) methylation, which has been linked with a good prognosis [13]. GSM biomarkers with possible therapeutic consequences concern EGFR, CDKN2A, BRAF, PTEN, and NF1[9]. MGMT promoter methylation is more common in GBM than in primary GSM and a tendency of increased survival in patients with hypermethylated MGMT promoter by improving the effectiveness of TMZ treatment was mentioned [61, 62].
The NF1 gene was found to be changed in 30% of GSMs due to indels. In human GBM tumors somatic mutations in the NF1 gene have been observed [56,57], amongst which splice site, non-sense, mutations, missense alterations, and frameshift indels were present. A number of the mentioned mutations have been recorded as germline changes in patients with neurofibromatosis, therefore are probably inactivating [47, 50]. It has also been recorded confirmation of the interaction between NF1 and GSM patients. Pathological events, such as p53 increased expression, indicate that exists no overexpression of EGFR, as in primary GBMs, and that the proliferation indices increase could result in a poor prognosis, in general [63]. In GSM cases have been revealed NF1 molecular changes via deleterious mutations and copy number losses. The function loss of NF1 increases RAS activity, inducing RAS/RAF/MEK/ERK pathway activation. MEK inhibitors as a single agent (PD0325901 and AZD6244) have been found to be efficient against a NF1-deficicient GBM cells subset dependent on RAF/MEK/ERK signaling [63]. In GSM the frequency of NF1 mutations was found to be 18% [56]. In a GBM mesenchymal type the NF1 gene is frequently deleted, however the NF1 gene deletion/mutation overall frequency was estimated to be almost 30% in mesenchymal GBM cases [57], similar to GSM specimens. It is important to notice that the PTEN frequency mutations or NF1 alterations were much greater in GSMs than in GBMs, stated to be 41% for PTEN and only 10% for NF1 in GBM cases [57]. (Table 2).
| Gene | Type of alterations | Protein alterations [8] |
| TP53 | Mutation | C135F, C238Y, H193R, H179Y, D281G, L111P, I255N, K132R, R175H, P80Lfs*43, R248Q, R248W, R273C, R282W, R342*, T125M, V272M, S241F, Y205H, V73Wfs*50, |
| PTEN | Mutation/Copy Number Alteration | Noncoding mutations appearing at hotspots C228T and C250T |
| TERT promoter | Mutation | C71Y, N184Efs*6, G36R, R130*, R130Q, R173C, G230*, N48K, L325P, S229*, R233*, V166Sfs*14, V175M, W274*, X55_splice, X268_splice, deep deletion |
| NF1 | Mutation/Copy Number Alteration | E1264*, I1679, Q2589*, Y2285Tfs*5, P1847Qfs*16, Y1680del, R1534*, R2637*, Deep DELETION |
| RB1 | Mutation/Copy Number Alteration | H733Ffs*13, S149*, R467*, M484Vfs*8, S567*S576Rfs*34, deep deletion |
| EGFR | Mutation/Copy Number Alteration | A289V, R222C, amplification |
| BRAF | Mutation | G32_A33dup, G466E, V600E |
| CDKN2A | Copy Number Alteration | deep deletion |
| CDKN2B | Copy Number Alteration | deep deletion |
| APC | Mutation | A735V, R876Q |
| STAG2 | Mutation | G935Vfs*2, K906Nfs*11, M318R |
| MSH6 | Mutation | L1244dup, T1133A |
| CBL | Mutation | R420L, R718* |
| SOX2 | Copy Number Alteration | Amplification |
| PTPN11 | Mutation | G60R, N308D, S502L |
| CREBBP | Copy Number Alteration | A1603T, deep deletion |
| ARID2 | Mutation | I124T, T1180K |
| FBXW7 | Mutation | R465H, R465C |
| SUZ12 | Mutation | G42Afs*30, T596Nfs*6 |
Table 2: Targetable alterations in GSM-The top19 genes in GSM
The amplification of EGFR was observed in 4% of GSM compared with 35% of GBM cases [9,49,51]. GSMs have an explicit genetic profile, similar to GBMs except for the amplification of EGFR [46] and recent data suggested that genes amplification on proximal 12q could facilitate a sarcomatous genotype development [52]. Until now, limited information is available regarding the epithelial component’s molecular genetics, observed in connection with GBM or GSM cases. Previous research showed the same standard of TP53 mutations in astrocytic and in epithelial differentiation areas of GBMs [64]. That finding has been initially detected for glial and mesenchymal regions in GSMs [59].
GSM, does not carry EGFR mutations or amplifications at the same frequency, indicating that may exist extra/alternative mechanisms driving carcinogenesis and eventually mesenchymal transformation into a sarcomatous phenotype. Similar studies observed a very low EGFR amplification prevalence in GSM, however they also showed frequent chromosome 7 (72%) gain containing the EGFR locus. It is important to notice that EGFR mutation or amplification is not surely demanded for EGFR activation. It remains unclear whether the activation of EGFR signaling pathway is present in GSMs cases, however maybe it is present thru not direct mechanisms and not surely thru overexpression of EGFR caused by gene amplification [65,66]. Previous researches showed a very low or absent amplification/overexpression of EGFR in GSM cases [12,46], but the recent copy-number analysis utilizing CNV microarrays revealed frequent EGFR amplification [18]. Other GSMs genomic analyses have detected EGFR amplification frequencies of 4% in a survey of 22 samples, as mentioned, and 74% EGFR gain in another one of 18 samples with one sample expressing EGFR amplification [46,67]. EGFR is considered to be a key oncogenic driver in GBM, amplified in 35-45% of IDH wild type GBMs [66].
In an inclusive whole-genome copy number analysis of GSM, a report showed that amplification of EGFR was unusual, but also showed frequent chromosome 7 gains, which contain the EGFR locus, among other genes comprising PDGF-A, CDK6, and c-Met [67]. It is not clear whether the EGFR pathway is indirectly activated in GSM thru other mutations.
EGFR mutations- targeted treatments are not expected to be essential therapeutic options in GSM due to genetic alterations low frequency. The EGFR amplification rate has been estimated 35-45% in IDH-wild-type GBMs [32], whereas in general, the alterations of EGFR are rare in IDH-mutated GBM but more dominant in IDH-wild-type GBM [68]. Although, mutations/ indels of EGFR have not been recorded in GSMs, the amplification of chromosome 7 (the region where the EGFR gene is located) was very frequent and appeared in 40% of GSMs, however in the EGFR locus no focal amplification was detected [6,46].
In GSM cases the DNA copy number losses were frequent. The main part of copy number loss concern chromosomes 9 and 10, regions comprising CDKN2A and CDKN2B genes. The CDKN2A gene encodes for proteins p16 and p14arf, which are tumor suppressor genes and regulate the p53 and RB1 cell cycle components (https:// www. omim. Org/entry/ 600160 # mapping). The CDKN2B gene encodes for the p15ink4b protein, a p16ink4 (CDKN2A) family member, and a cell growth regulator that inhibits G1-phase progression (https:// www.omim.org/entry/ 600431?search =cdkn2b&highlight = cdkn2b). CDNK2A loss was frequent in 35-60% of IDH-wild type GBM cases [55,69,70], and CDKN2A homozygous loss was also frequent in GBM cases (35-50%) (International Agency for Research on Cancer, 2016), whereas in a GSMs micro-array study, CDKN2A homozygous loss was detected in 14 of 18 (77,7%) GSM specimens examined [67]. (Table 1).
The molecular etiology which is involved in the transformation of GBM into GSM remains unclear. The progress in GSM has been associated with alterations in signaling pathways, such as MAPK (TP53, EGFR, and RASGRF2), phosphatidyl-inositol/calcium (CACAN1s, ITPRs, and PLCs), and focal adhesion/ tight junction (PTEN and PAK3) pathways [18,49]. In GSM the mesenchymal transformation has been associated with TWIST1, SNAI2, and MMP-2/MMP-9 up-regulation [71]. According to the WHO the TWIST, SNAI2, MMP2 and MMP9 expression is a typical element of mesenchymal regions, indicating epithelial to mesenchymal transition (EMT), and may play an essential role. The molecular alterations revealed in GSM were comprised 219 cases [62], and was found lower frequency of EGFR copy number amplification (CNA) in GSM (8%) versus GBM (up to 50%), and also was recorded that prior case series estimating both the glial and the sarcomatous components of GSM showed that both components shared common genetic and chromosomal alterations of the conventional GBM, findings which suggest a common clonal origin for both components [62].
The increased expression of PD-1 and PD-L1, is an EMT evidenced element in a diversity of tumors [72-74]. The PD-1/PD-L1 role in the pathogenesis of GBM and the potential for targeting the mentioned pathway has been examined [75]. Elevated levels of those proteins and of tumor infiltrating lymphocytes have been detected in GSMs versus GBMs in a series comprising 233 WHO Grade IV gliomas with 9 GSM cases [76].
Other possible pathways involved in GSM concern the OX40L/OX40 pathway activation, which it is responsible for strong immunity and antitumor effects in GBM cases [77]. The loss of DNA copy number was present in regions coding for diverse OX40L/OX40 pathway protein ingredients, such as NF-kB, NF-kB2 (p52), NF-kB2 (p100), PKC-theta, Perforin, IKK-alpha, and Calcineurin A (catalytic). Inversely, various regions that displayed chromosome loss were locations which coded for WNT pathway proteins Tcf (Lef), Dickkopf-1(DKK1), TCF 7 L2 (TCF4), beta-TrCP, Sirtuin 1, and BMI-1. Most of those WNT pathway proteins in an over-expressed or activated status, with the DKK1 exception, promote cell survival and proliferation [78]. Nevertheless, DKK1 is regarded to be a negative regulator of WNT signaling pathway, and has been implicated as a candidate gene in medulloblastoma as it is epigenetically silenced [79], whereas its loss, may result in the activation of the WNT signaling pathway with subsequent cell survival and proliferation. It has also been observed that DKK1expression resulted in glioma cell sensitivity to chemotherapy-induced apoptosis [80].
NF-kß, a protein complex which is responsible for DNA transcription controlling, is able to in-duce cell proliferation and antiapoptosis in case of improper regulated or constitutively activated. It has been recorded that NF-kß abnormal activation in GBM, led to cell invasive abilities, radio-therapy resistance, and even the promotion of mesenchymal phenotype [81].
Although the multifactorial role of NF-kß is involved in a biological processes various number, such as cell survival and proliferation, motility, DNA repair, inflammation, etc., a direct path-way which leads to GBM pathogenesis is ambiguous. In GSM cases it has been detected a copy number loss in the region encoding for NF-kß, indicating that the NF-kß pathway activation seems not to have a critical role in GSM pathogenesis. Nevertheless, it is possible that loss of NF-kß could result in DNA repair mechanisms loss, leading to neoplasia [82].
The role of BRAFV600E mutation in GSM cases is controversial according to previous reports [83,84]. Activating BRAF-V600E mutations have been frequently observed in cases of pediatric glial and glioneuronal brain neoplasms [47,48]. However, it has been reported that BRAF V600E mutation is present in 10% of GMS cases, compared with 3% of GBMs, whereas SOX2 gene amplifications and mutation of MSH6 are present about in 10% and 20% of GBM cases, respectively [85,86]. Moreover, Zaki et al., compared common gene changes, greater than 5%, in GSM, GBM, and soft tissue sarcoma, and among those, GSM shared only four genes with GBM, none with sarcomas, whereas nine common genes were found monadic to GSM amongst the 5% threshold for each respective tumor type [9]. They also reported that BRAF mutations (V600E protein alteration, G32A33 duo, G466E), SOX2 amplification (11%), and MSH6 mutations (L1244dup, T11 33A protein alteration), were special to GSM [9]. Previous studies recorded that most of those mutations overlap with GBM and other cancer types, however, GSM carries its own genetic mutations, such as, Suppressor of Zeste 12 (SUZ12), and Box and WD Repeat Domain Containing 7 (FBXW7) [5,9,13,51,86,87]. (Table 2).
TGF-β superfamily signaling is responsible for a broad spectrum of cellular functions both in normal and tumor growth, as is implicated in determining the mesenchymal stem cell differentiation pathway [88,89] and in the EMT regulation in lung cancer and mesothelioma cases [90,91]. TGF-β/BMP signaling plays an essential role in osteoblast-genesis and bone formation [92, 93]. TGF-β signaling pathway also activates down-stream SMADS, CTNNB1, MYC and FOS signaling pathways, which could result in the malignant induction of a pro-neural-mesenchymal transition in that tumor by increasing the following mesenchymal transcriptional factors expression, SNAI1, SNAI2, TWIST1, and ZEB1 [71,94]. Those transcription factors are responsible for re-programming and activate the mesenchymal signature transcription in the recurrent GSM tumor [18,71,91]. Consequently, TGF-β signaling seems to play a critical role in modulating mesenchymal stem cell lineage selection and imposed the mesenchymal differentiation progression into the osteo-genic lineage by controlling the main transcription factor’s expression and activities [71,88].
The main noticeable difference between GSMs and GBMs concerns the collagen gene signature, indicating a more mesenchymal-like and extra-cellular matrix rich micro-environment. Collagens type I (COL1A1, COL1A2), III (COL3A1) and VI (COL6A2, COL6A3) were highly up-regulated in GSM cases [49]. The collagen-signature is involved in the gene’s groups functional analysis which differentially expressed between GBMs and GSMs, as “focal adhesion” is one of the distinguishing groups. Especially, COL6A3 seems to be a reliable marker of GSM tumors, as its expression was increased in the sarcomatous element, whereas it was practically absent in the gliomatous one [49].
Gene’s overexpression which are associated with integrin complexes ITGA5-ITGB1-CAL4A3 and ITGB1-NRP1 in GSM cases when compared with GBMs showed that GSMs were more invasive and migratory tumors, as the mentioned integrins were implicated in the EMT processes [95,96]. A remarkable translocation between RABGEF1 and GTF2I RD 1P1 genes was revealed in three GSM samples. The close proximity of the mentioned genes is able to result in a possible long deletion or read-through transcript, as the distance between two fused RNA fragments is nearly three kb long. The RABGEF1 gene last exon is fused with the GTF2IRD1P1 gene, leading to its inactivation. As the RABGEF1 gene was associated with some cancers development that alteration may play a significant role in GSM development, however further research is required to explain the mentioned translocation impact [97,98].
The current viewpoint concerning the GSM cellular origin maintains the monoclonal theory that both glial and mesenchymal components may be come from a common neoplastic neuro-ectodermal precursor cell [46,99,100]. From a histological point of view, gliomatous and sarcomatous components of GMSs share specific genetic alterations and possibly come from a common clonal origin [31,59]. The analysis of gliomatous and sarcomatous elements of eight GSM cases by comparative genomic hybridization after micro-dissection detected that both components shared 57% of the discovered chromosomal imbalances. Nevertheless, the chromosomal alterations number in GSMs was significantly lower than that in GBMs, suggesting a greater genomic stability in GMSs [59]. Other authors described that gliomatous and sarcomatous elements of GSM shared common genetic alterations and chromosomal imbalances of the type conventionally described in GBM [52]. Those alterations comprised gains on chromosomes 7,9q, 20q, and X, and losses on chromosomes 10, 9p, and 13q. GSMs were also recorded to have a fewer chromosomes number implicated in imbalances, indicating a genomic stability greater level in GSMs [52]. It has also been recorded those chromosomes 9 and 10 showed the highest number of losses, and the copy number of gains mainly appeared on chromosome 7 in GSM samples [67]. The loss of LOH on 10q is a common genetic alteration in primary and secondary GBM, indicating that 10q may comprise tumor suppressor genes [101]. In GSM, LOH 10q was also frequently observed (88%) [31].
GSM is a rare clinicopathological entity, and is difficult to differentiate from GBM on clinical information, however shows a genomic and molecular aspect distinct from GBM and soft tissue sarcoma, even though is classified by the WHO as a GBM variant. The current review demonstrated that most GSM tumors have somatic alterations of PIK3/Akt (PTEN, PI3K) and RAS/ MAPK (NF1, BRAF) signaling pathways which are essential for tumor development and therapy resistance. GSMs, regarding somatic alterations, are considerably similar to GBMs, with a greater NF1 and PTEN alterations frequency, more similar to frequencies detected in mesenchymal GBMs. A better understanding of the cellular and molecular profiling of GSM and the development of targeted therapies may help individuals affected by this enigmatic tumor. In the meantime, early diagnosis and a multidisciplinary approach to treatment remain crucial against GSM and may improve further survival.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
