
Case Report | DOI: https://doi.org/10.31579/2690-4861/487
1Department of Internal Medicine, Resident Physicians at Mather Hospital at Northwell Health in Port Jefferson, NY
2Department Transitional Year, Resident Physicians at Mather Hospital at Northwell Health in Port Jefferson, NY
3Department Internal Medicine, Resident Physicians at Mather Hospital at Northwell Health in Port Jefferson, NY
*Corresponding Author: Salman Syed, D.O., Resident Physician at Mather Hospital at Northwell Health, Port Jefferson, NY.
Citation: Salman Syed, Hadassah Stein, Fareeha Aajmal, (2024), Mixed autoimmune hemolytic anemia during recovery from Immune Thrombocytopenia (ITP): A Case Report, International Journal of Clinical Case Reports and Reviews, 18(1); DOI:10.31579/2690-4861/487
Copyright: © 2024, Salman Syed. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 03 June 2024 | Accepted: 07 June 2024 | Published: 18 June 2024
Keywords: antibodies; red blood cells; diagnostic; clinical presentation
Mixed autoimmune hemolytic anemia (AIHA) is characterized by hemolysis with the coexistence of warm and cold autoantibodies. It is a relatively rare condition, accounting for 6.5-8.3% of AIHA cases. It is diagnosed by identifying IgG and C3d with cold agglutinins on monospecific direct antiglobulin testing. We present a unique case of a 69-year-old female who developed mixed hemolytic anemia while she was being treated for chronic idiopathic thrombocytopenic purpura (ITP).
A 69-year-old female with a history of ITP and anemia was sent to the emergency room by her hematologist to receive a blood transfusion for low hemoglobin found in outpatient labs. It was associated with fatigue, generalized weakness, and shortness of breath. On review of systems, she denied having any abnormal bleeding, bruising, dark stools, fever, chest pain, palpitations, paroxysmal nocturnal dyspnea, abdominal pain, urinary symptoms, rash, or joint pain. Additional medical history included hypertension, hyperlipidemia, anxiety, depression, and acid reflux. Home medications included 60 mg oral prednisone daily and Romplostin weekly for ITP, along with Metoprolol, Omeprazole, and Paroxetine for her other chronic medical conditions. She did not use tobacco or recreational drugs and did not drink alcohol.
Vital signs revealed a heart rate of 110 but were otherwise unremarkable. The EKG showed sinus tachycardia. During the physical exam, the patient did not appear distressed. However, her skin appeared pale, she had scleral icterus, and she was tachycardic on auscultation.
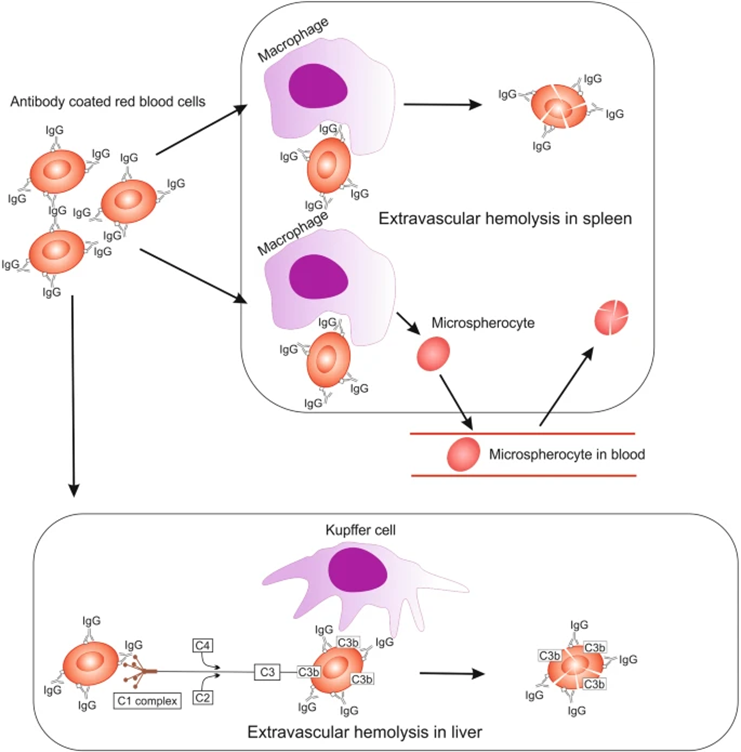
Figure 1: Hemolysis during warm autoimmune hemolytic anemia (WAIHM): Antibody-coated red blood cells pass through the spleen, where they are engulfed and degraded by macrophages. Some erythrocytes will become microspherocytes due to losing some cell membranes and being released back into circulation. Microspherocytes have rigid cellular membranes and lack flexibility, so they ultimately are re-sequestered in the spleen and degraded. Alternatively, IgG-coated red blood cells travel to the liver, where weak activation of complement leads to extravascular hemolysis [2]
Source http://creativecommons.org/licenses/by/4.0/
Labs showed a leukocytosis of 34.93k/ul, platelet count of 557k, hemoglobin of 6.6g/dl, and hematocrit of 18.4%, which subsequently dropped to 5.4 g/dl and 15%, respectively, on repeat blood draw in the ED. The complete metabolic panel showed normal values for Blood urea nitrogen (BUN), aspartate aminotransferase (AST), and alanine aminotransferase (ALT) but elevated total Bilirubin of 6.1 with a direct bilirubin of 0.4. Blood sugar was elevated at 183, which was expected given steroid usage. The chest radiograph was negative for any acute pulmonary process. The patient was COVID-negative. She received one unit of packed red blood cells in the emergency room and was admitted for further workup.
Further hematologic workup showed lactate dehydrogenase (LDH) of 690 (135-214 microliter) and haptoglobin less than 20 (30-200 mg/dl) during admission. Immunofixation electrophoresis (IFE) and serum protein electrophoresis (SPEP) testing for plasma cell dyscrasias were negative. The Direct Antiglobulin Test (DAT), also known as the Direct Coombs Test, was positive for C3d and warm antibodies. Cold agglutinin titer was greater than 1:512 (normal is less than 1:64). Abdominal ultrasound revealed splenomegaly without hepatomegaly. Lower extremity duplex was negative for clots, and the echocardiogram showed an ejection fraction of 77% with normal right ventricular systolic function and
elevated right ventricular pressure, suggesting moderate pulmonary hypertension. Based on the results, the patient was diagnosed with mixed-type AIHA.
Follow up investigation was then conducted to elucidate the cause of AIHA. Peripheral
blood smear showed reticulocytosis of 22% (normal range 0.5 to 2.5%) with a normal estimated platelet count of 460k, which was expected given that the patient was already on home steroid treatment. Thrombophilia testing and hepatitis serology were negative. Serology for HIV, Syphilis, CMV, and Mycoplasma pneumonia were likewise negative, while EBV IgG was positive with negative IgM. CT scan of the chest and pelvis done to rule out a lymphoproliferative process demonstrated splenomegaly but was otherwise negative. Bone marrow biopsy was obtained and analyzed with cytogenetic karyotyping negative for gross chromosomal abnormalities and fluorescent in situ hybridization (FISH) panel study negative for chromosomal rearrangements characteristic of myeloproliferative disorders and myelodysplastic syndromes. She received four units packed-RBCs during admission and was discharged on a steroid taper, Romploistosm, and Rituximab once Hb was stable at 8.5g/dl.
Mixed warm/cold autoimmune hemolytic anemia is a relatively rare condition, and current evidence on its optimal management is limited. Additionally, the variability in diagnostic criteria across sources further complicates both the diagnosis and management of this pathology.[1]
This is a unique case of mixed warm and cold autoimmune hemolytic anemia (AIHA) emerging in a patient with pre-existing ITP. The patient's diagnosis was confirmed by a positive Direct Antiglobulin Test (DAT) for both IgG and complement C3d, along with a high cold agglutinin titer[2,3,4]. The patient's elevated lactate dehydrogenase (LDH) and low haptoglobin levels were consistent with a hemolytic process. The splenomegaly noted on ultrasound and CT was consistent with splenic sequestration and destruction of red blood cells. An echocardiogram was performed to assess cardiac function given the anemia. Chronic iron deficiency anemia can lead to hyperdynamic circulation and hypoxic pulmonary constriction with resultant pulmonary hypertension, as was revealed on this patient’s echocardiogram [5]. She was treated with steroids, romiplostim, and rituximab, which helped stabilize her hemoglobin levels.
Mixed AIHA involves the simultaneous presence of warm and cold autoantibodies and is relatively rare, accounting for 6.5-8.3% of AIHA cases [1]. Approximately half of diagnosed cases have no identifiable cause [1]. Warm autoantibodies, primarily IgG, typically react at body temperature and lead to extravascular hemolysis via splenic macrophage-mediated destruction [3]. Cold agglutinins, usually IgM, react at lower temperatures, triggering complement activation and intravascular hemolysis[ 6,7,8].
The coexistence of warm and cold antibodies can lead to a more severe anemia and can present diagnostic and therapeutic challenges [9,10,11]. The diagnostic workup includes comprehensive hematologic and serologic testing to rule out secondary causes such as lymphoproliferative disorders or infections. Standard treatment is with steroids, romiplostim, and rituximab.
Implications for Practice
This patient was found to have mixed AIHA in the setting of known ITP and anemia. This case cautions against anchoring, especially in a complex clinical context. Anchoring is a cognitive error where excessive weight of information obtained early on impedes one’s ability to change a presumed diagnosis in the face of new knowledge [12]. In this case, the patient’s acute anemia could have been attributed to their underlying ITP and chronic anemia if thorough investigation had not been pursued.
One limitation of this case is the absence of a clear underlying cause for the mixed AIHA as investigations for secondary causes such as infections, lymphoproliferative disorders, and other autoimmune diseases yielded negative results. However, it is not unusual for cases of AIHA to be idiopathic [13,14]. Although lymphoproliferative disorder was a differential in the setting of the patient’s splenomegaly, the bone marrow biopsy came out to be negative. Additionally, the patient was not followed for a lengthy interval at the time of this publication, so the long-term prognosis and potential for relapse in this patient with multiple hematologic comorbidities were not determined. [15,16]
This case report contributes to the limited literature on mixed AIHA by detailing the clinical presentation, diagnostic algorithm, and treatment approach for patients with concurrent ITP. The complexity of managing such cases necessitates a thorough and multidisciplinary approach. Continued documentation and analysis of similar cases will enhance understanding and improve management strategies for this rare clinical presentation.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
