
Review Article | DOI: https://doi.org/10.31579/2693-7247/061
Touro College of Pharmacy. 230 West 125th New York, NY, 10027, United States.
*Corresponding Author: Iva Srdanovic, Department Touro College of Pharmacy. United States.
Citation: Iva Srdanovic (2022) Late Catch-up In-Stent Restenosis and Stent Thrombosis for 2nd Generation Biodegradable Polymer Drug Eluting Stents J. Pharmaceutics and Pharmacology Research. 5(3); DOI: 10.31579/2693-7247/061
Copyright: © 2022, Iva Srdanovic, This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 29 November 2021 | Accepted: 01 December 2021 | Published: 18 January 2022
Keywords: Stent Restenosis; Stent Thrombosis; Biodegradable Polymer.
Second generation drug-eluting stents (DES) have reduced the rate of in-stent restenosis (ISR) to < 10% [1, 2] in patients undergoing PCI. However, late catch-up in-stent restenosis remains an issue despite the advent in drug-eluting stent technology from 1st to 2nd generation drug eluting stents. The late catch-up phenomenon has been associated with serious consequences and adverse events and remains an important issue in modern practice, despite medical advances with more biocompatible and biodegradable polymers, as well as new therapeutic agents and thinner scaffolds in the development of 2nd generation DES polymer systems. The idea behind biodegradable polymers has been to provide a more controlled elution profile for DES drugs, allowing for long-term therapeutic, but sub-toxic, drug levels in the arterial tissue to prevent ISR and stent thrombosis. However, the late catch-up phenomenon remains an issue resulting in 2ndary revascularization beyond the first year of primary PCI, as it is defined as ISR >1 year. Late stent thrombosis (<1 year) and very late stent thrombosis (>1 year) also remain an issue for biodegradable polymer systems. A long-term presence of the polymer can cause inflammation and thrombogenesis, which is dependent upon polymer degradation kinetics in case of biodegradable polymer DES systems. However, the cause of late catch-up restenosis and stent thrombosis is multi-factorial from a drug-in-polymer formulation perspective of drug release kinetics, e.g., drug elution profile, drug physiochemical properties, and polymer degradation kinetics. It appears that the focus should be on controlling burst release for both late catch-up restenosis and stent thrombosis phenomena.
A Review on the release kinetics (burst release) influence on the late catch-up phenomenon and stent thrombosis for 2nd generation biodegradable DES systems
Drug eluting stents release an anti-proliferative drug from a polymer matrix coating metallic struts to prevent arterial smooth muscle cell (SMC) proliferation and neointimal hyperplasia at the site of vessel injury upon stent implantation. A stent is composed of metallic struts and polymer coating containing the anti-proliferative agent as either a reservoir or polymer matrix formulation. Late catch-up restenosis is defined as in-stent restenosis beyond 1 year of primary PCI procedure. [3] It is caused by the narrowing of the stented segment as a result of inefficacious concentrations of the anti-proliferative agent in the arterial tissue necessary to inhibit SMC proliferation and neointimal hyperplasia. One of the major challenges in DES performance has been developing an optimal release profile to keep drug concentration at efficacious but sub-toxic levels in the arterial tissue over a sufficiently long enough period of time to prevent smooth muscle cell proliferation and subsequent restenosis without inhibiting re-endothelization, as stent thrombosis has been associated with inhibited re-endothelization of arterial tissue. In other words, drug elution profile should be sustained for a long enough time-period to provide anti-restenotic effects, especially until the polymer degrades in case of biodegradable polymers.
Drug release kinetics characterized by initial burst release have been associated with late catch-up restenosis and stent thrombosis. A rapid, uncontrolled initial release of drug and rapid release rate may cause tissue toxicity without sustaining efficacious and required drug therapeutic levels in the arterial wall long term due to systemic loss of the therapeutic agent. At the same time, this burst release leaves the tissue exposed to polymer known to cause inflammation and, thus delays vascular healing and inhibits re-endothelization [4,5]. In other words, burst release results in a high loss of drug to the systemic circulation without it being retained in the arterial tissue for a long enough time to exhibit its anti-proliferative effects and inhibit late catch-up restenosis. [4,5]. Burst release can subject arterial tissue to more drug than it can absorb and retain [6], so high and extreme drug doses overwhelming tissue receptors can cause augmented fibrin deposition, intra-intimal hemorrhages, mural thrombus, medial necrosis and excessive arterial expansion, all associated with stent thrombosis and exacerbated neointimal tissue [7,8].
Drug physiochemical properties play an important role in burst release and release kinetics in general, as well as drug uptake and retention by the surrounding arterial tissue, when the drug is being released from the polymer matrix. Polymer properties, such as hydrophilicity, degree of crystallinity, pore size and pore density, influence drug diffusion through polymer matrix and polymer degradation kinetics, as well as subsequent drug release rate. Understanding formulation factors influencing burst release and release kinetics, as it pertains to the drug type, polymer degradation kinetics, as well as certain coating techniques associated with burst release, can help explain the late catch-up restenosis phenomenon and stent thrombosis in biodegradable drug eluting stent systems.
Factors Influencing Burst Release:
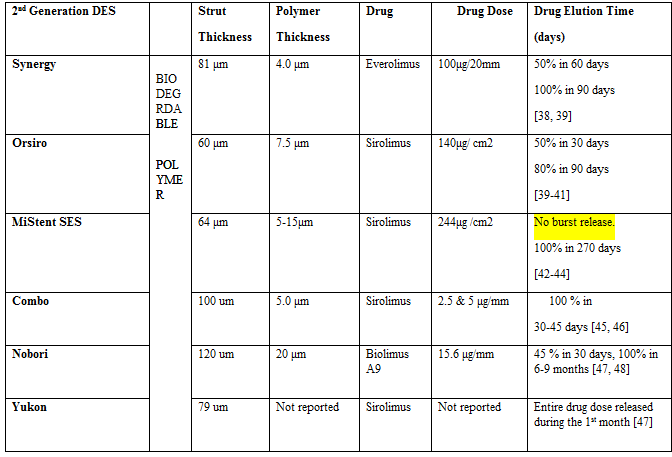
Drug physiochemical characteristics controlling drug elution profile, drug concentration in polymer matrix, polymer permeability and thickness for both DES reservoir and polymer matrix systems, solvent evaporation rate following DES coating, as well as polymer properties governing degradation mechanisms and drug release kinetics are all factors contributing to burst release [9-19]. Initial rapid release of the drug from DES systems should not result in high depletion of the polymer matrix, because such release would not allow for longer elution profiles at the site of action in the arterial tissue following stent implantation, necessary to inhibit SMC proliferation and prevent late catch-up restenosis [20, 21]. Burst release results in high drug doses released initially and lost to systemic circulation, leaving the arterial tissue void of efficacious therapeutic levels of the anti-proliferative agent necessary to inhibit ISR [4,5]. At the same time, drug overwhelming tissue receptors could cause toxicity, increasing the risk of inflammatory and thrombogenic response, which is exacerbated by the fact that the arterial wall remains subject to polymer reactions for several months until its degradation. Table 1. shows different biodegradable FDA approved stents, the initial burst release followed by the rest of the elution profile. Figure 1. shows the time it takes for the polymer to degrade. Even though burst release can be favorable to heal the initial impact and injury of stent implantation to the arterial tissue, uncontrolled burst release represents a major challenge in developing an optimal DES drug release profile to reduce the incidence of late catch-up restenosis and stent thrombosis in biodegradable DES polymer systems.
Drug Physiochemical Properties Affecting Drug Elution Profile and Burst Release: -Limus Family of Drugs - Lipophilicity, Ionizability, and Crystallinity
Sirolimus and its derivatives have been the anti-proliferative agents of choice for 2nd generation biodegradable DES systems. -Limus family of drugs are cytostatic immunosuppressants with a wide therapeutic window, leaving arterial tissue viable as opposed to causing cell death, which is the case with cytotoxic agents like paclitaxel used in 1st generation DES systems. Sirolimus and its derivatives inhibit the cell cycle at the G1 to S phase. Sirolimus and its analogues, such as zotarolimus (Endeavor, Medtronic) and everolimus (Xience, Abbott), have varying degrees of lipophilicity and ionizability. Drug lipophilicity/hydrophobicity, ionization/polarity (pKa values) have an influence over drug elution profile from the DES polymer matrix into the aqueous interstitial fluid and blood plasma [physiological pH (7.37-7.43)] in terms of drug dissolution kinetics as well as drug diffusion through and solubility in the polymer reservoir or matrix DES system. Zotarolimus is extremely lipophilic, and the high degree of lipophilicity and poor water solubility slow down the dissolution profile in the interstitial fluid of arterial tissue; thus, increasing dissolution kinetics to increase bioavailability should deliver just enough as opposed to too much drug with better permeability through cell membrane. The slow dissolution profile results in negligible drug tissue concentrations conductive to re-endothelization [22]. High degree of lipophilicity also allows for lower effective concentration at the site of action as compared to sirolimus in terms of reducing the incidence of adverse vascular events associated with restenosis [23, 24]. Zotarolimus has a shorter in vivo half-life than sirolimus, but the same high affinity binding to the immunophilin FKBP12 [25]. Biolimus A9 is another highly lipophilic derivative of sirolimus, readily absorbed by arterial tissue [26].
On the other hand, the more polar derivatives of sirolimus, such as everolimus, have a faster dissolution profile from the polymer matrix into the surrounding tissue media that can result in burst release in comparison to sirolimus and zotarolimus, which lack in ionizable groups. Everolimus faster dissolution profile is countered by a slower rate of cell uptake and subsequent lower tissue concentrations as compared to the more hydrophobic anti-proliferative agents, which prevents tissue receptors from being overwhelmed by high drug doses associated with stent thrombosis. It is important to note that drug transmural diffusivity and retention by arterial tissue, even though affected by the water/lipid avidity of therapeutic agents within the arterial tissue, has also been shown to be more influenced by the specific protein binding sites, dictating drug retention time within the arterial wall. Everolimus has a much higher interaction with mechanistic target of rapamycin complex 2 as compared to sirolimus, shorter half-life and better bioavailability [27], allowing for lower concentrations to be effective at the site of action.
Burst release is also a result of higher drug concentration being dispersed in the outer layers of DES polymer system - a limitation of different solvent-based coating techniques as opposed to high drug content in DES as evident in Table 1 [28-30]. An optimal DES formulation itself should have a lower drug content to prevent a high initial burst release. Low MW drugs have higher propensities for burst release as a result of osmotic pressure [31]. The -limus family of drugs represents a group of relatively small compounds with the following molecular weight: sirolimus (915 g/mol) [32], zotarolimus (966 g/ml) [33], everolimus (958 g/mol) [34] and biolimus A9 (986.3 g/mol) [35]. Drug solubility in DES polymer matrix systems also affects drug release kinetics from the formulation.
Degree of crystallinity is another factor contributing to burst release. Drug-eluting stents containing crystalline drug particles may be favored for better drug delivery over the conventional approach of spraying amorphous form in polymer solutions onto metallic struts especially when combined with biodegradable polymers and an anti-inflammatory agent like the -limus drug family agents that can counter any tissue reaction arising from presence of polymer degradation products [25, 36]. Amorphous drug elution profile from DES is dependent on diffusion along a concentration gradient, and thus associated with a rapid, uncontrolled drug burst upon initial release. On the contrary, crystalline drug elution is dependent on dissociation/dissolution reaction from the crystalline lattice associated with a high activation energy barrier, eliminating the initial burst release characteristic of diffusion-controlled mechanisms, and resulting in a more consistent and gradual drug release rate throughout polymer absorption regardless of the amount of drug remaining in the coating [4, 37].
MiStent containing sirolimus crystals embedded in a biodegradable polymer matrix composed of polylactide-co-glycolic acid (PLGA) showed a negligible burst release in comparison to conventional DES containing amorphous sirolimus drug, as well as a more linear, long-term drug elution profile extending over a 9-month time period. Conventional biodegradable DES are characterized by a relatively short, logarithmic-type pattern drug elution profile with an initial burst release, follow by the remaining drug being released from the polymer matrix and eventually falling below therapeutic levels with most of the drug dose lost to systemic circulation following burst release, while the polymer remains intact. This increases chances of late catch-up restenosis and stent thrombosis (Figure 1) [4]. Previously it has been shown that crystalline sirolimus elution from MiStent sustains higher drug loads in tissue compared to conventional coated stents with similar drug loads of sirolimus drug in amorphous form [20]. Drug elution without an initial
burst release, or a negligible burst release, lasting over 9 months for MiStent releasing crystalline sirolimus drug, can possibly be associated with lower rates of stent thrombosis and inhibited vessel restenosis.
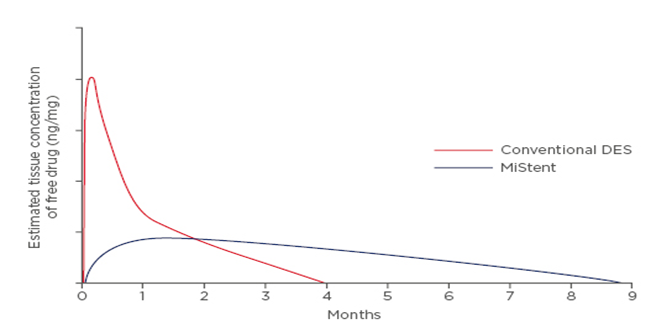
Drug release (yellow) and polymer absorption (blue) arranged by length of polymer degradation. Modified from source [47].
Release Mechanisms and Polymer Properties Affecting Drug Release Kinetics:
Drug release kinetics from biodegradable polymers are initially characterized by diffusion/dissolution and diffusion and/or swelling and then controlled by polymer degradation (bulk degradation and/or surface erosion). Swelling of polymer systems increases free volume and mesh size and is followed by drug diffusion through the swollen network into the site of injury caused by stent implantation. Polymer swelling can facilitate diffusion by increasing the aqueous solvent content in the formulation and creating pores for drug diffusion out of the matrix as it partitions between the polymer and the aqueous solvent depending on its polarity/solubility [49, 50]. Polymer degradation kinetics ultimately control drug release from DES biodegradable polymer systems, resulting in pore formation that eventually dictates drug release from the matrix, allowing for facilitated drug transport/diffusion and release as the polymer molecular weight decreases and the polymer chains become less entangled upon degradation, as is the case with PLGA polymer degradation and erosion [51]. Drug release is highly dependent on the following polymer properties: molecular weight, monomer composition, degree of crystallinity, porosity, hydrophilicity, degree of swelling and degree of cross-linking, as these govern drug diffusion and polymer degradation kinetics. Polymer degradation is also dependent on temperature and pH [52, 53-55]. These factors contribute to changes in polymer glass transition temperature (Tg) and, as a result affect the degree of crystallinity and water uptake, which determines polymer permeability, subsequent drug diffusion through the matrix, and polymer degradation kinetics. [56-58]. Particle shape and size also influences drug release, as the ratio of surface area to volume is an important parameter facilitating more water contact [52] with increasing surface to volume ratio resulting in accelerated polymer degradation kinetics [59]. Polymer surface morphology can also be modified to control drug release from polymeric DES systems [60].
Commonly Used Biodegradable Polymers: PLA, PGA, PLGA
Commonly used synthetic biodegradable polymers for DES polymer reservoir and matrix systems include thermoplastic aliphatic poly(esters) such as polylactic acid (PLA), polyglycolic acid (PGA), and poly (lactic-co-glycolic acid) (PLGA). PLA can be made using two types of monomers: two stereoisomers of lactic acid (D and L). PLA prepared using D-lactic acid, PDLA, is a crystalline material resulting from its regular chain length; on the other hand, PLA prepared using L-lactic acid, PLLA, has a semi-crystalline structure. PLLA and PDLA polymer blend yields PDLLA, which has amorphous characteristics. The presence of methyl groups on PLAs makes them more hydrophobic in nature [61]. Therefore, the chirality of the monomer influences drug release kinetics because it has an effect on the biodegradability and mechanical properties of PLA. Studies have shown that D and D/L forms of PLA degrade faster than the L form, as the latter is more crystalline in nature in comparison to the other two [62-66]. PLA degrades through ester bond backbone hydrolysis [67].
PLGA (Poly (d,l-lactic-co-glycolic acid) is a copolymer of lactic acid (PLA) and glycolic acid (PGA) whose physical, thermal and mechanical properties depend on the ratio and type of the monomers used for the polymer blend [68]. PLGA properties can be modified changing the molecular weight and PGA to PLA copolymer composition to adjust the degree of crystallinity, hydrophilicity, and glass transition temperature (Tg)of the copolymer blend. Increasing the ratio of lactic to glycolic acid can increase polymer hydrophobicity with an increase in methyl groups, which in turn reduces the rate of water penetration through the device and subsequent hydrolysis and degradation, to allow for a more controlled drug release profile. As a rule, higher glycolic acid content yields faster degradation rates due to PGA being more hydrophilic as compared to PLA allowing for higher rates of water uptake and hydrolysis of PLGA ester linkages. 50:50 PGA to PLA ratio is the exception to the rule because it exhibits the fastest degradation rates with increasing PGA content leading to faster polymer degradation kinetics below 50% [55, 69, 70 ].
Biodegradable polymers like PLA, PGA and PLGA, degrade by bulk erosion associated with burst release and subsequent loss of the anti-proliferative agent too early on during the arterial healing process, as well as inhibition of re-endothelization associated with stent thrombosis [50, 71-73]. Drug lost to systemic circulation as a result of the initial uncontrolled burst release results in sb-therapeutic levels in the arterial tissue needed to prevent SMC proliferation and neointimal hyperplasia and subsequent late catch-up restenosis long-term. At the same time, synthetic PLA, although biocompatible, can take more than a year to degrade and therefore carries a risk of late and very late stent thrombosis [49, 74]. In case of Orsiro, it takes up to 15 months to degrade and is, therefore, present long after the drug has been eluted in the first three months and lost to systemic circulation [47]. Additional problems may arise from poor mechanical performance and generation of acidic products from polymer degradation, which may lead to inflammatory responses and induce neointimal hyperplasia and subsequent restenosis, as well as thrombosis at lower pH values for the biological environment [75, 76]. The acidic by-products of PGA as well as its fast degradation kinetics make it an unfavorable candidate to be used for as a single polymer matrix and can lead to local inflammation at acidic pH as a degrading component of a PLGA polymeric system [77].
Bulk Erosion vs Surface Erosion: Implications for Burst Release for Biodegradable DES Polymer Systems
Bulk erosion, characteristic of PGA, PLA and PLGA degradation, is associated with more burst release and unpredictable drug release profiles as compared to surface erosion; it is a suboptimal mechanism for controlled drug delivery [73]. Biodegradation by surface erosion means that a polymer goes through a heterogeneous process degrading from the surface inward proportional to the surface area, while maintain its bulk integrity, so the drug release form surface eroding systems is often correlated with a controllable and reproducible erosion rate with thicker systems having longer erosion times. In other words, polymers degrade quickly at the surface without the penetration of water molecules by hydrolysis [69, 71, 73]. Contrary to surface erosion, in bulk erosion, the rate at which the water penetrates the device is higher than the rate of erosion, so water penetration rate exceeds the rate at which the smaller polymer constituents are converted into water-soluble materials, resulting in homogenous degradation of the entire matrix with an initial burst release [71, 73]. As mentioned earlier, PLA, PGA and PLGA degrade by bulk erosion associated with burst release [50, 71-73] and unpredictable drug release rates-a suboptimal mechanism for drug delivery as compared to surface erosion [73].
Solvent Evaporation Rate and Burst Release:
Coating techniques involving solvent evaporation upon drug deposition onto the polymer/scaffold or scaffold alone can result in burst release. Complications associated with solvent-based coating techniques, such as dip coating and spray coating, include bridging, pooling and lack of uniformity, which is especially the case with coating thickness less than 0.5 mm [78, 79]. Varying concentrations of drug and polymer can result in burst release with an uneven drug distribution such that the outer layers are rich with the drug of choice in comparison to the rest of the matrix [11, 28-30]. Multiple rounds of spray coating can lead to significant mixing between different layers of the polymer matrix [80] and result in higher drug content near the surface [81].
Drug release kinetics, especially burst release, have been associated with late catch-up restenosis. An initial uncontrollable release of the DES therapeutic agent can lead to sub-therapeutic levels of the antiproliferative agent in the arterial tissue, which is needed at optimal levels over an extended time-period to “safeguard” the tissue from narrowing in; most of the drug is released initially as part of burst release and lost to systemic circulation shortly after, so the drug concentration falls below the efficacious therapeutic levels. Burst release has also been associated with delayed re-endothelization and subsequent stent thrombosis. Drug overwhelming tissue receptors at high initial doses released as part of burst release can cause augmented fibrin deposition, intra-intimal hemorrhages, mural thrombus, medial necrosis and excessive arterial expansion, all associated with stent thrombosis and exacerbated neointimal tissue. Biodegradable polymers are especially prone to burst release as a result of bulk erosion mechanism for polymer biodegradation. Drug physiochemical properties, as well as polymer properties governing polymer degradation kinetics, also influence the drug elution profile and subsequent drug concentration in the arterial tissue that needs to be sustained long-term to prevent late catch-up restenosis, but at subtoxic levels to avoid stent thrombosis. When too much anti-proliferative agent is released too soon, then the drug is lost to systemic circulation, leaving the tissue exposed to struts and polymer without a drug concentration efficacious enough at inhibiting SMC proliferation, which essentially leads to late catch-up restenosis (ISR>1 year). The focus of DES technology should be optimal release profiles of the anti-proliferative agent with special attention on burst release to improve the device for better clinical outcomes.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
