
Case Report | DOI: https://doi.org/10.31579/2578-8949/067
Department of dermatology CHU Hassan II, FES ,Morocco.
*Corresponding Author: Elharrouni Alaoui A, Department of dermatology CHU Hassan II, FES, Morocco.
Citation: Elharrouni Alaoui A, Baybay H,Chaoui R, Douhi Z, Elloudi S, Mernissi FZ.(2020) Kindler syndrome: clinical particularities about two cases reports in two siblings. Journal of Dermatology and Dermatitis.5 (2); Doi:10.31579/2578-8949/067
Copyright: © 2020 Elharrouni Alaoui Aicha, This is an open-access article distributed under the terms of The Creative Commons. Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 17 March 2020 | Accepted: 30 March 2020 | Published: 01 April 2020
Keywords: Keywords
The Kindler syndrome, the fourth major type of hereditary epidermolysis bullosa (HEB), is a rare autosomal recessive genodermatoses, characterized by trauma-induced blistering, cutaneous atrophy, and progressive poikiloderma, in association with mucosal inflammation. We report tow new sibling cases of this rare syndrome.
Kindler’s syndrome(KS) is a rare type of genetic skin condition belonging to the class of bullous poikilodermia.It is characterized by skin fragility and blistering at birth followed by development of marked photosensitivity and progressive poikilodermatous skin changes in later years.We report two new sibling cases of this rare syndrome.
This condition was seen in two girls aged 4 and 8 years, born of a first-degree consanguineous mariage with Kindler syndrome of varying degrees of severity.There are presented to us with a history of formation of multiple blisters, predominantly over the acral sites since birth. The one of patient also complained of photosensitivity.
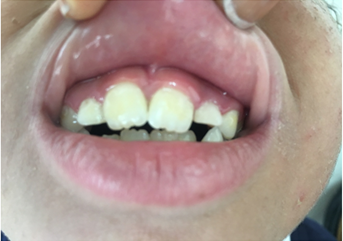
Dermatological examination the older sister revealed a well‑defined hemorrhagic flaccid bulla over dorsolateral aspect of the foot. Multiple hypopigmented scars with cigarette paper wrinkling were present over the dorsa of feet, hands, and elbows .there also had poikilodermia of gradual onset makes hypopigmented and hyperpigmented macules with telangiectasia, and atrophic scars over the face with angular cheilitis and gingival hypertrophy(figure :a-b-e).
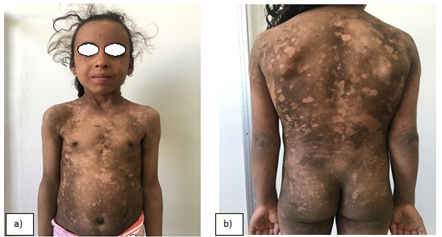
And Cutaneous examination in younger sister revealed multiple hypopigmented and a few hyperpigmented macules of variable sizes, distributed over his face, neck, trunk, and limbs .Poikilodermatous skin changes were present. The overall texture of the skin was xerotic with marked cutaneous atrophy. The palms showed hyperkeratosis with diminution of palmar creases. Skin over the hands and neck was dry, atrophic and photosensitive to the sunlight. The patient was short stature (figure : c-d). Clinical diagnosis and genetic study were in favor of Kindler syndrome

Kindler syndrome (KS) is a rare autosomal recessive genodermatosis, characterized by skin fragility and blistering at birth followed by development of marked photosensitivity and progressive poikilodermatous skin changes in later years(1). KS is associated with mutations in the KIND1 (FERMT- 1) gene that result in either hypomorphic,or complete loss of Kindlin-1 activity(1).Clinical findings in KS are increased skin fragility, acral blistering, photosensitivity, atrophy, and poikiloderma. The photosensitivity usually decreases over time, coinciding with decreased blister formation by 10‑12 years of age, although some degree of photosensitivity usually persists even after that age(1-2). Poikiloderma, and atrophy generally occur at sun‑exposed sites, but it can also present at non‑exposed sites. palmoplantar keratoderma, pseudosyndactyly, leukokeratosis of lips and oral mucosa, pseudoainhum, xerostomia, phimosis, dental caries, periodonitis,Gingivitis and periodontitis are also prominent features(2-3). Gastrointestinal symptoms, including constipation and severe colitis, can also occur in KS. Diagnosis of Kindler syndrome is based upon clinical evidence. Electron microscopic examination is used in particular to rule out congenital bullous epidermolysis(4). Detachment of layers at two or three different levels in relation to the dermal-epidermal junction described in the literature forms a specific but inconsistent feature of Kindler syndrome(2). The management of KS is largely symptomatic, encompasses protective measures that include sun protection and care of acral bullae and wounds by appropriate topical and systemic antibiotic treatment(2-5).
Kindler syndrome is a rare syndrome. Its diagnosis is pratically clinical. Different levels of cleavage have been described in electron microscopy. We have reported two new observations of two sisters who borned as a first-degree consanguineous marriage in a family whose clinic and genetic study were the keys to diagnosis.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
