
Case Report | DOI: https://doi.org/10.31579/2641-0419/255
1 Department of Paediatric Cardiology, Klinikum Großhadern, Ludwig-Maximilians-University, Marchioninistraße 15, 81377 Munich, Germany.
2 Department of neonatal care, Klinikum Großhadern, Ludwig-Maximilians-University, Marchioninistraße 15, 81377 Munich, Germany.
3 Department of inborn errors of metabolism, Dr. von Hauner Children's Hospital, Ludwig-Maximilians-University, Lindwurmstr. 4, 80337, Munich, Germany.
4 Department of Pediatrics, Dr. von Hauner Children's Hospital, University Hospital, Ludwig-Maximilians-Universität München, Munich, Germany.
*Corresponding Author: Fiona Becker-Dettling, Department of Paediatric Cardiology, Klinikum Großhadern, Ludwig-Maximilians-University, Marchioninistraße 15, 81377 Munich, Germany.
Citation: Fiona B. Dettling., Robert D. Pozza, Adelheid Kley, Esther M. Maier., Stephanie R. Vandewiele. et all (2022). Infantile Mitochondrial Cardiomyopathy – A Case Report Presenting a Rare Cause for Hypertrophic Cardiomyopathy. J. Clinical Cardiology and Cardiovascular
Interventions, 5(5); DOI:10.31579/2641-0419/255
Copyright: © 2022 Fiona Becker-Dettling, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 23 February 2022 | Accepted: 15 March 2022 | Published: 22 April 2022
Keywords: infantile hypertrophic cardiomyopathy; mitochondriopathy; mitochondrial respiratory chain complex v deficiency; mitochondrial atpase 6; mitochondrial atpase 8; m.8528n>c; case report; neonates
Infantile hypertrophic cardiomyopathy (HCM) comprises a group of rare and heterogeneous diseases and is the second most common cause of cardiomyopathy in childhood and adolescents. In general, the muscular hypertrophy results in histological and functional disorder. The prognosis depends on the underlying aetiology. However, HCM represents a high risk of mortality especially in neonates. Rarely, mitochondrial dysfunction is accountable for HCM. Here we describe a case of a rapidly progressive infantile HCM caused by complex V deficiency due to overlapping mt mutations in ATP6 and ATP8 with lethal course in a premature neonate suggesting early genetic testing in risk groups to clarify differential diagnosis, and determine thereby prognosis and rational therapy options.
Mitochondriopathy
The term primary mitochondriopathy is a generic term for diseases with impairment of mitochondrial function[1]. Primary mitochondrial disorders have been estimated to have a minimum birth prevalence of 1 in 5000 [2]. Usually, these diseases take on divers clinical characteristics and multisystemic damage is seen due to inherited or sporadic mutations. These mutations affect either nuclear or mitochondrial genes [3]. Mitochondria are highly dynamic organelles found in all eukaryotes and are normally maternally inherited. Pathophysiologically, diverse posttranslational features (e.g., protein-protein interaction, transducing, enzymatic function) can be impaired and can lead to decreased energy production, with detrimental effects in many organs. The final common pathway of aerobic energy production proceeds via the ATP synthase, located at the inner mitochondrial membrane[2]. The ATP synthase is also known as the complex V of the respiratory chain and consists of one hydrophobic membrane-bound and one hydrophilic cytosolic component. The hydrophobic inner subunits are encoded by the mitochondrial genes ATP6 and ATP8. Consequently, complex V mutations result in critically impaired oxygen exploitation and ATP deficiency [4]. Mostly tissues dependant on high energetic levels such as brain, central nervous system, heart and kidneys are affected and display various symptoms. Recent data has shown that myocardial mitochondrial impairment presents at a young age and consequently leads to rapid cardiac dysfunction [5]. Cardiomyocytes not only show a high turnover metabolism, but they also effect important intracellular processes as calcium and electrolyte homeostasis. Consequently, the increased production of free oxygen radicals leads to massive cellular oxidative stress and myocardial damage ultimately resulting in heart failure. This form of heart failure may result in early death of the child [6].
The definition of hypertrophic (obstructive) cardiomyopathy includes left ventricular wall thickness independent from pathologic left ventricular filling pressureshttps://leitlinien.dgk.org/files/33_2014_pocket_leitlinien_hypertrophe_kardiomyopathie.pdf; German guideline. The heart structures affected the most are the interventricular septum and the ventricles with a z-score > 2. Regarding its epidemiology, HCM is considered as a rare disease due to an incidence of 4.7 per 1.000.000 children. Recent data showed a two-year mortality of 30% for infants younger than one year [7]. An early clinical onset often reveals an aggressive disease by causing left ventricular outflow obstruction, diastolic dysfunction, myocardial ischemia, and mitral regurgitation as well as electrical conduction problems [8]. Infants who suffer from HCM may show a wide variety of symptoms e.g. tachydyspnoea, edema, drinking weakness, failure to thrive or may even be asymptomatic [9]. The underlying etiology can be multi-causal. Differentials mostly include genetic variants such as mutations in sarcomeric protein genes, RASopathies (e.g. Noonan-Syndrome), inherited errors of metabolism as glycogen/lysosomal storage diseases (e.g. hemochromatosis, Fabry disease, Pompes disease, M. Niemann-Pick), fatty acid oxidation disorders, malformation syndromes, neuromuscular/neurologic (e.g. Friedreich's ataxia, Duchenne/Becker muscular dystrophy, neurofibromatosis, tubular sclerosis), endocrine or autoimmune dysfunctions (e.g. diabetes mellitus, diabetic fetopathy, pheochromocytoma, acromegaly, lupus erythematodes, polyarteritis nodosa); [7]. Rarely, mitochondrial disorders have also been reported. Although up to 60 % of HCM patients are diagnosed with an autosomal dominant mutation, hypertrophy is not always genetic and can even be iatrogenic (e.g. consequences of antimetabolite therapy, anthracycline, cyclophosphamide, drugs, anabolic steroids) or may result from malnutrition or nutrient deficiency (e.g. carnitine and selenium)[3].
Patient information:
History: The female newborn was prenatally diagnosed with HCM and growth retardation. The patient was born with 34 + 0 weeks of gestation by caesarean section after an otherwise uneventful pregnancy. She was the first child of a 36-year-old 2nd gravida, 1st para. Notably, the maternal patient history also displayed an unspecified hypertrophic cardiomyopathy. The maternal chromosomal genetic testing showed 46, XX. Exome sequencing showed a heterozygous mutation of uncertain significance in MYBPC3 (VCV000180411). The fetal genetic analysis including chromosomal screening, FMR1-Repeat-diagnostic and cardiomyopathy panel did not reveal any pathologic findings.
Perinatal course: Neonatal key data: Apgar score: 9 / 9 / 10 at 1 / 5 / 10 minutes. NA-pH: 7.29. Weight: 1200 g (<1st>
Diagnostics:
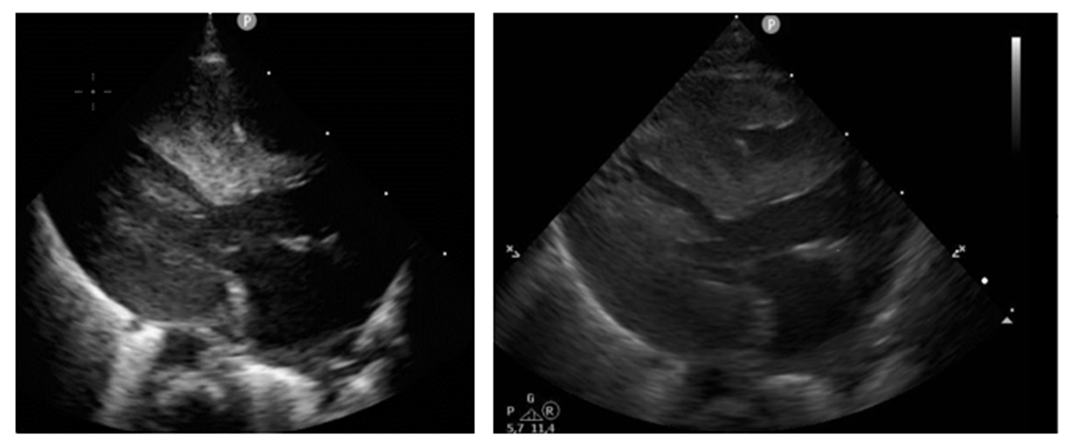
A chest X-ray displayed the upper mediastinum and cardiac shadow to be widened in supine position with massive cardiomegaly. There were no pleural effusions, no evidence of circumscribed infiltrates, no pneumothorax (see Fig. A). Postnatal echocardiography showed a situs solitus with atrioventricular-ventriculoarterial (AV-VA) concordance and confirmed the prenatal findings of biventricular, especially left-side concentric myocardial hypertrophy. There was no left ventricular outflow tract obstruction (LVOTO), the biventricular systolic function overall was sufficient with diastolic function impaired. The right-sided structures were unremarkable. No patent ductus arteriosus (PDA), only a small atrial septal defect II (ASD II, 5mm) with left-right shift (LRS) were seen. The septum was 0.941 mm (see Figure. B1 and B2). The routine sonography of the head and abdomen showed normal findings consistent with age and were otherwise unremarkable. Because the mother's vaginal swab was positive for Streptococcus agalactiae and Candida glabrata, the infant received a prophylactic antibiotic therapy with piperacillin/tazobactam for 48 h and antimycotic prophylaxis with fluconazole.

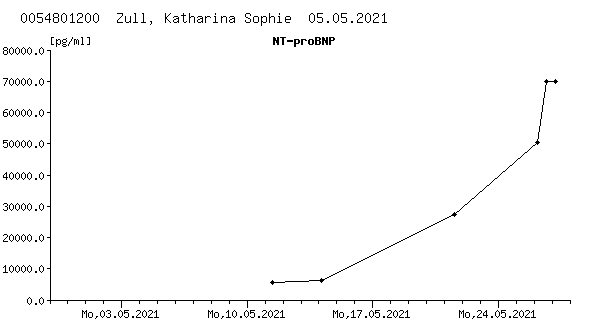
Further course: In the further course of 10-14 days the infant adapted well and was stable in room air. Because of prematurity an oral respiratory stimulation with caffeine was continued since the first day of life. As we saw tachypnoea, she received respiratory support using HFNC for two days. Since birth, POCT analysis showed an intermittent mild lactatemia (4-6 mmol/l) with a steadily rising trend, simultaneous blood glucose level was low (min. 34 mg/dl). Echocardiographically, we saw a significant increase of the biventricular hypertrophy. Furthermore, a severe diastolic dysfunction was seen. Due to increased pro-NT-BNP (max. 6144 pg/ml) and the progressive diastolic heart failure an anticongestive treatment with propranolol (starting dose 2 mg/kg/d) p.o. was administered on the 9th day of life (see Figure. C1 and C2) . Drug administration was done under glucose monitoring which showed no adverse events or symptoms of hypoglycaemia. After 24h, the lactate dropped to 2.8 mmol/l. Due to recurrent arterial hypotension, a dose reduction to minimal 0.6 mg/kg/d propranolol was necessary. Consecutively, we saw a rising trend of the lactatemia. Therefore, we slowly increased slowly the dose without any complications but nevertheless, lactate even more increased (see Figure. C1). An adjuvant oral therapy with coenzyme Q and carnitine was given supplementarily. In the event of lactatemia as well as intermittent arterial hypotension, multiple volume administrations with crystalloid solutions were given. Diuresis was timely and sufficient at all times.

The infant got oral nutritional support on the first day of life with breast milk and premature infant formula. Additional partial parenteral nutrition was necessary because of intermittent hypoglycaemia until the 20th day of life (independently from propranolol). In addition, she got oral supplementation of vitamins A, D and K as well as iron as standard treatment for prematurity. Due to insufficient oral intake, a probing of meals was implemented. The meconium passage was protracted despite repeated irrigation. When lactate levels got high, only parenteral glucose was given.
Differentials: In general, possible causes of HCM include congenital metabolic defects, genetic syndromes (e.g. Noonan syndrome) or mitochondriopathy. Upon admission in neonatology intensive care unit, additional diagnostic testing was engineered: Concerning metabolic aetiology a general metabolic screening including amino acids, acyl carnitine in plasma and organic acids in urine were found as unremarkable, likewise the neonatal screening. Remarkable was a mild elevation of ammonia (114 µmol/l) on the fifth day of life and persistent high levels of lactate, nonexplicable for merely cardiac cause Genetically, for a verification/exclusion of Noonan’s syndrome a target genetic testing questioning PTPN-11 and RAF1 was implemented. Additionally, whole-exome sequencing (WES) for mother and patient was performed early to clarify differentials.
Decline: At 14th day of life (35+6 SSW), there were positive signs of systemic infection (IL-6 135 pg/ml). A respiratory support with HFNC was re-implemented (6 l/min, FiO2 0.23). Several blood cultures were made, a PICC catheter removed and she was given a single shot vancomycin. At this point, during general deterioration and a rapid lactate increase to 9.6 mmol/l with exhausted respiratory compensation, the patient was transferred to the IMC paediatric cardiology unit for further therapy. Echocardiography demonstrated a rapidly progressive biventricular hypertrophy without obstruction with progredient signs of severe diastolic dysfunction. In the following hours, the patient became clinically unstable - sepsis with multi organ failure was suspected. In addition, the labs showed leukopenia (4.3G/l), the lactate was 8.1 mmol/l). An empiric standard antibiotic therapy was started and later intensified with meropenem and vancomycin i.v. Suddenly the patient became bradycardic, showed respiratory exhaustion, and required resuscitation due to circulatory failure. She was intubated and given surfactant as radiological findings revealed a severe ARDS. After one hour of cardiopulmonary resuscitation, stable circulation was achieved with adrenalin infusion at 0.37 μg/kg/min. Volume boluses were necessary several times. Nevertheless, peripheral blood pressures could only be measured intermittently, pulses were only weakly palpable. During and after resuscitation the lactate increased to max. 28 mmol/l. Despite successful resuscitation, a satisfactory hemodynamically stable situation could not be established under persistent catecholamine therapy. The poor prognosis including the lack of curative therapeutic options were intensively discussed with the parents and compassionate care was decided. The next day, the small patient became hemodynamically instable. She was extubated on her father's arm and died peacefully.
Postmortem: Microbiologic findings revealed blood cultures positive for Staphylococcus epidermidis, Staphylococcus aureus and Klebsiella pneumonia. Antibiotic sensitivity to the administered antibiotics was given. Concerning genetics, the specific testing for Noonan syndrome resulted in unremarkable findings. The WES confirmed a mitochondriopathy by demonstrating a point mutation (GRCh37.p13: chrMT: m.8528N >C) affecting the overlapping genes for ATP6 and ATP8. The consequence of this mutation is a start-loss of ATP6 (ENST00000361899: c.2N>C; p.Met1Thr) and a missense in ATP8 (ENST00000361851: c.163N>C; p.Trp55Arg). This variant is described as pathogenic (ClinVar ID: 9640) and was found in all 65 sequencing reads obtained for this position in the patient. This variant was also detected in the exome data of the mother, but displaying 2 wild type counts (out of 86 reads) explaining the obvious milder phenotype. In summary of all findings and events, the circulatory insufficiency occurred in the context of sepsis, aggravated by congenital hypertrophic cardiomyopathy with underlying mitochondriopathy. Unfortunately, no cardiac biopsy could be performed after death to perform tissue based mitochondrial analysis.
With this case of an infantile mitochondrial cardiomyopathy, we presented a rare cause for HCM. Due to the impact on high energy depending tissues, cardiac involvement is frequently seen in mitochondrial disorders [10]. About 50% of patients with mitochondrial affliction are also diagnosed with cardiomyopathy [11]. Especially in childhood, some of the cardiac complications need early implantation of a cardioverter-defibrillator or a pacemaker [12] or even cardiac transplantation in monosymptomatic disease. The failing cardiac condition is characterized by a pathologic histology and function of the heart muscle due to defect in the respiratory chain within the mitochondria [13]. The mitochondrial DNA (mtDNA) is composed of a circular DNA structure of ~16,569 base pairs and encodes 13 proteins of the respiratory chain as well as 2 rRNAs and 22 tRNAs. The remaining mitochondrial genes are mostly nuclear encoded and are transferred on a protein basis into the mitochondria [14]. The mtDNA is almost exclusively maternally inherited [10], genetic variants can be present as „ homoplasmic“, meaning that the complete genetic material of the mitochondria contains this variant or as „heteroplasmic“ if only a certain percentage carries a given variant [15]. In this case, the genetic analysis detected an overlapping double ATP synthase defect (ATP6/ATP8) resulting in an altered amino acid sequence und dysfunctional ATP exploitation. The specific mutation (m.8528T>C) was previously reported, additionally linked to hyperammonemia [16], with poor overall outcome resulting in the patients’ death likewise within the first months of life [20], highlighting the dramatic impacts of this mutation. In our case, we only saw mild signs of hyperammonemia, but could find the variant in both the mother and the patient: The mother is heteroplasmic for the m.8528N >C variant and the daughter was homoplasmic resulting in a more severe and finally lethal phenotype. This highly emphasizes the mutation correlates with disease and highlights the importance of early and possibly additional genetic testing of the index person’s relatives to workup differential diagnosis [17].
The most common sign in mitochondriopathies is the presence of lactemia. Increased levels of lactate are the result of induced stress responses including hyperactivated metabolic rate, sympathetic nervous system activation, accelerated glycolysis and a modified bioenergetic supply. Higher levels of lactate correlate with poor prognosis and non-survival, too [18]. Additionally, cardiac dysfunction is found in >40% of sepsis patients and enhances mortality rates [19]. In our case, lactatemia as well as high levels of pro-BNP were present since birth and showed a rapidly rising trend in accordance with the simultaneous increase of the biventricular hypertrophy additionally worsening the situation. The adjustment of anticongestive treatment only shortly improved the situation and could not prevent the fatal course. Even if the genetic diagnostic possibilities and understanding in the diverse pathophysiological background as well as therapeutic opportunities have developed the last years, there is still no curative therapy existing so far and there is still an urgent need for novel targeted prevention and curative therapy.
Infantile hypertrophic cardiomyopathy displays a genetically heterogeneous disorder with significant high morbidity and mortality, especially when caused by mitochondriopathy. As we showed with this case, early genetic analysis coupled with detailed phenotyping should be considered in patients with positive familiar history for HCM, persisting lactatemia, nondescript metabolic screen and unclear fatal course with the aim to clarify differentials and diagnosis. Furthermore, we suggest an early interdisciplinary cooperation of all relevant subspecialties as neonatologists, pediatric cardiologists, metabolism experts and geneticists to determine prognosis and rational therapy options without delay. In this case, with a known diagnosis before fatal outcome, a change in treatment goal towards palliative care would have been desirable. Therefore, early diagnosis und prognosis assessment would allow improvement of treatment strategies and precise genetic counseling with regard to further pregnancies.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
