
Research Article | DOI: https://doi.org/10.31579/2692-9406/135
1Department of Pediatrics, University Clinics of Kinshasa.
2Department of Pediatrics, MonkoleHospital Center.
3Department of Pediatrics, HGR Kintambo.
4Department of Epidemiology, School of PublicHealth.
5Monkole Hospital CenterLaboratory.
*Corresponding Author: Ariane Keto, Ph.D., Department of Pediatrics, University Clinics of Kinshasa.
Citation: Keto MA, Tshilolo L, Budiongo NA, Mfulani G, Mafuta E. et al (2022) Impact of G6pd Deficiency on Sickle Cell Disease in Children in Kinshasa Hospitals: a Case-Control Study. J. Biomedical Research and Clinical Reviews. 7(3); DOI:10.31579/2692-9406/135.
Copyright: © 2022 Ariane Keto, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 09 November 2022 | Accepted: 16 December 2022 | Published: 23 December 2022
Keywords: G6PD deficiency; sickle cell disease; child and kinshasa
Background and aim: Glucose-6-Phosphate Dehydrogenase (G6PD) deficiency and sickle cell disease are two genetic diseases of the red blood cell that both cause hemolytic anemia. The objective of this study is to determine the clinico- biological impact of G6PD deficiency on sickle cell disease in childrenin Kinshasa.
Materials and method: This is a case-control analytical study of 103 G6PD-deficient sickle cell patientsand 309 non-G6PD- deficientsickle cell patients. Analysis of G6PD activitywas performed by ELISA, and hemoglobin electrophoresis by capillaris to confirm sickle cell disease.For all children, sociodemographic, clinical and biological variables were analyzed.
Results: The mean age of sickle cell deficient and non-deficient patients was 9.82±4.5 years and 9.48±3.8 years respectively. There were slightly more female sickle cell deficient patients (55.2%) while in the non- deficient group, there was a male predominance (51.8%) but no statistically significant difference (p>0.05). All morbid events occurring in sickle cell patients were greater in the G6PD deficiency group (p˂0.05). Evaluated antecedent complications occurringin sickle cell patients wereassociated with G6PD deficiency (p˂0.05),except for stroke.No statistically significant difference was noted in physicalsigns between sicklecell deficient and non-deficient groups(p>0.05). The hemogramshowed no difference between the two groups in steady state and the hemolysis markers evaluated also showed no significant differences in steady state (p>0.05) between the two groups.
Conclusion: G6PD deficiency aggravates the acute clinical manifestations and complications of sickle cell disease withoutaffecting the occurrence of stroke. And has no impact on the hematological parameters of sickle cell disease children in stationary phase.
Sickle cell disease and Glucose-6-Phosphate Dehydrogenase (G6PD) deficiency are two genetic diseases of the red blood cell mainly described in Sub- Saharan Africa and in particular in DRC. These two abnormalities are responsible for hemolytic anemia [1]. The genes involved are located on different chromosomes and therefore these two diseases are transmitted independently although they may be associated. Some authors believe thatG6PD deficiency has little or no impact on sickle cell disease while others believe that it worsens the clinical and biological profiles of sickle cell disease [2].
This comorbidity has been the subject of several scientific publications without showing an increase in the number of hemolytic crises, infectious episodes, and transfusions and chronic complications, or a lack of influence on the severity of the disease in such a combination [3-5]. Some authors believe that G6PD deficiency would have a protective effect since the deficiency of enzymatic activity could contribute to the premature disappearance of red blood cells, which would result in the release of a new generation of young circulating cells. Piomelli et al. in Kenya in 1972 found that these two anomalies seemed to attenuate each other: sickle cell disease made the enzyme deficiency less apparent to the point that the deficient genotype seemed to be masked [6].
Several authors have found that G6PD deficiency does not worsen the clinical and biological status of sicklecell patients; these include Firemponget al. in Ghana in 2016 and Diop et al. in Senegal in 2005 [3,7]. Others on the other hand have found that G6PD deficiency worsens the diseasestate of sickle cell patients: including: Bernaudin et al, in France in 2018, Benkerou et al., in France in 2013, Lohit et al , in India in 2019, Mounkaila et al , in Niger in 2015 and Bouangaet al , in Congo in 1998 [2, 8-11].
These different studies sufficiently demonstrate the high prevalence of the comorbidity of G6PD deficiency and sickle cell disease mainly in Africa. In the DRC, no study has been conducted on this comorbidity, yet it is one of the countries most affected by G6PD deficiency. Moreover, it is a country located in the sickle cell belt of Central Africa where sickle cell disease and malaria endemically [12, 13]. In addition, there is a high frequency of self- medication, particularly with antimalarial drugs, which are oxidizing molecules, exposing to acute hemolytic crises in case of G6PD deficiency (Quinine, Chloroquine, Mefloquine, Halophantrine) [14-19].
Thus, given the high prevalence of sickle cell disease - G6PD deficiency comorbidity and the high frequency of self-medication, we found it appropriate to undertake the present study, the objective of which is to determine the clinic-biological impact of G6PD deficiency on sickle cell disease among children in Kinshasa.
Nature, setting and period of study
This is a case-control study, which consisted of determining the clinico- biological impact of G6PD deficiency on sickle cell disease in children recruited as an active thread in medical facilities in Kinshasa. It covered the period from June 2017 to June 2019, a duration of 2 years.
Population and sampling
Study hospitals were selected by convenience sampling. The consecutive selection method, in active file, was used to identify the children to be submitted to the inclusion criteria. The first group consisted of homozygous G6PD- deficient sickle cell children confirmed by hemoglobin electrophoresis or molecular testing, aged 6 months to ≤18 years of age recruited in the active file, and followed up as an outpatient in the selected hospitals, and the second group consisted of age- and sex-matched nondeficient sickle cell children recruited in the same setting as the first group, i.e., one case for every three controls [20].
Variables of interest
The variables evaluated based on the clinical and biological manifestations of sickle cell disease were sociodemographic data (age, gender, origin),clinical variables (the notion of coca cola urine emission, the number of previous transfusions, the number of previous vaso-occlusive crises, the number of infectious episodes, the history of complications: priapism, aseptic necrosis of the femoral head, notion of stroke, notion of malleolar ulcer, physical signs) and biological data (determination of G6PD enzyme activity, complete blood count, total bilirubin) [12-14].
The samples were analyzed at the laboratory of the training and health support center (CEFA) and the Monkole hospital Center.
Conduct of the study
All children included in the study were identified using medical records and contacted to participate in the study. Children who met the inclusion criteria were first given a clinical examination that included a history and physical examination. The elements collected during the clinical examination made it possible to set up an ad hoc questionnaire. Acute sickle cell crises were assessed by the average annual number requiring emergency admission over the past 2 years (14). A physical examination, including weight measurement using a scale while undressed; height measurement using a horizontal scale in a supine or standing position according to age, with feet together.
Then, we took blood samples from each child. A 5 ml sample of venous blood was taken in two tubes; one dry for biochemical analysis, and the other with anticoagulant (EDTA) to carry out the hemogram, the determination of G6PD enzymatic activity by the ELISA method as well as hemoglobin electrophoresis; These tubes were stored at 4°C until the analysis was performed and a blood sample was taken on Whatman filter paper with conventional FTATM cards for G6PD. The samples collected were coded and immediately sent to the CEFA and Monkole Hospital laboratories.
Data processing and analysis
All data were collected and entered on Epi info 7, transported to Excel 2010spreadsheet, analyzed with SPSS for Windows version 22.0, and presented in tables and graphs.
Continuous quantitative variables with a normal distribution were presented as means ± standard deviation; those with an anomalous distribution, as median (interquartile range). Categorical variables were described as absolute and relative frequency (%).The comparison of the means or medians of the two groups was made, as appropriate, using the Student’s T test or the Mann Whitney U test; the Pearson Chi-square test, the Fisher's exact test or the trend test were used to compare the proportions. The factors associated with G-6-PD deficiency were sought using the logistic regression test. The odds ratio and the 95% confidence intervals were represented and allowed to measure this association. For all the tests used, the threshold of statistical significance was p less than 0.05.
Ethical considerations
The protocol of this work was submitted to the Ethics Committee of the School of Public Health and obtained approval under the number ESP/CE/028/2019. This work was conducted in strict compliance with the fundamental principles of ethics. Informed consent was obtained from the parents or guardians of the children by means of a written form after explanation of the purpose and implications of the study, the observations to be made, and the benefits and risks for the child.
Sociodemographic characteristics of the study population Clinicalinformation of the study population Clinical profile of G6PD deficient and non-G6PD deficient children with sickle cell disease biological information Factors associated with G6Pd deficiency
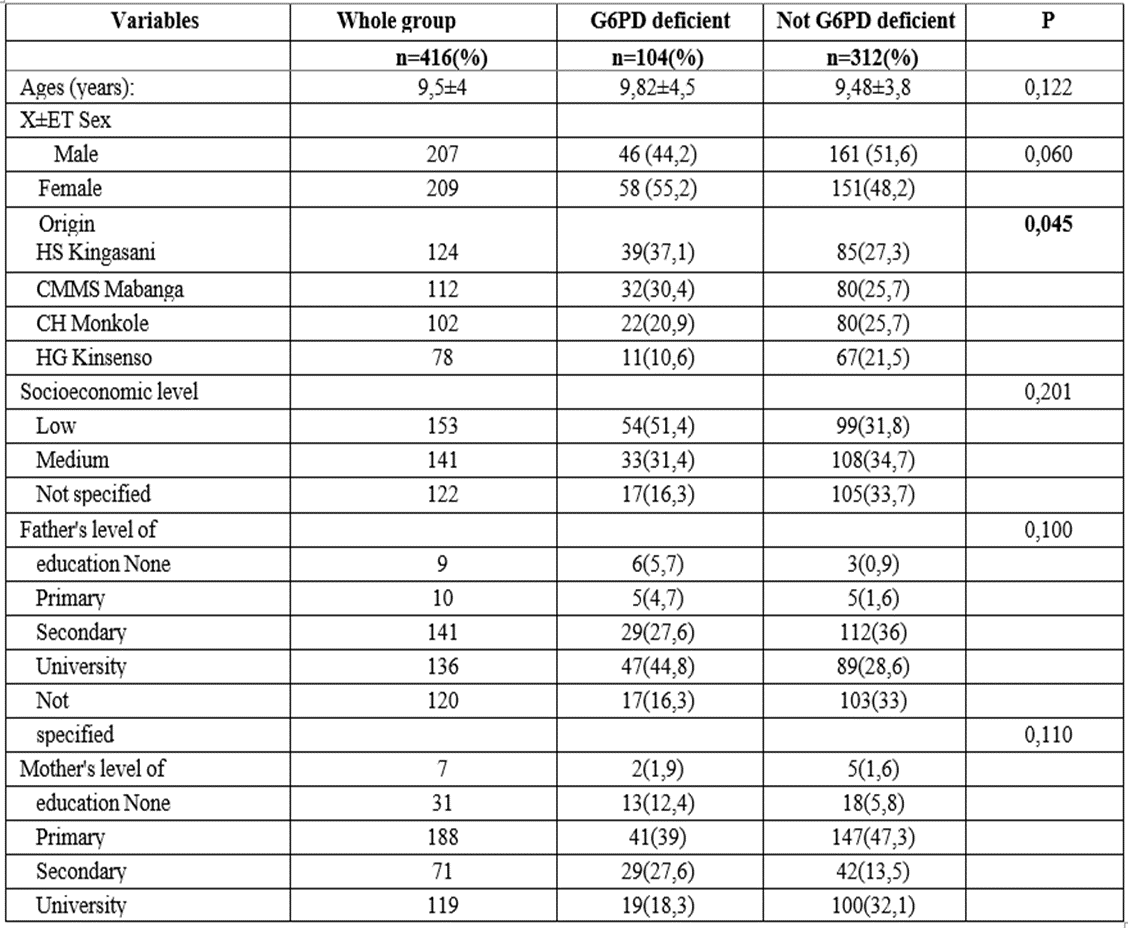
Table 1: Description of the study population
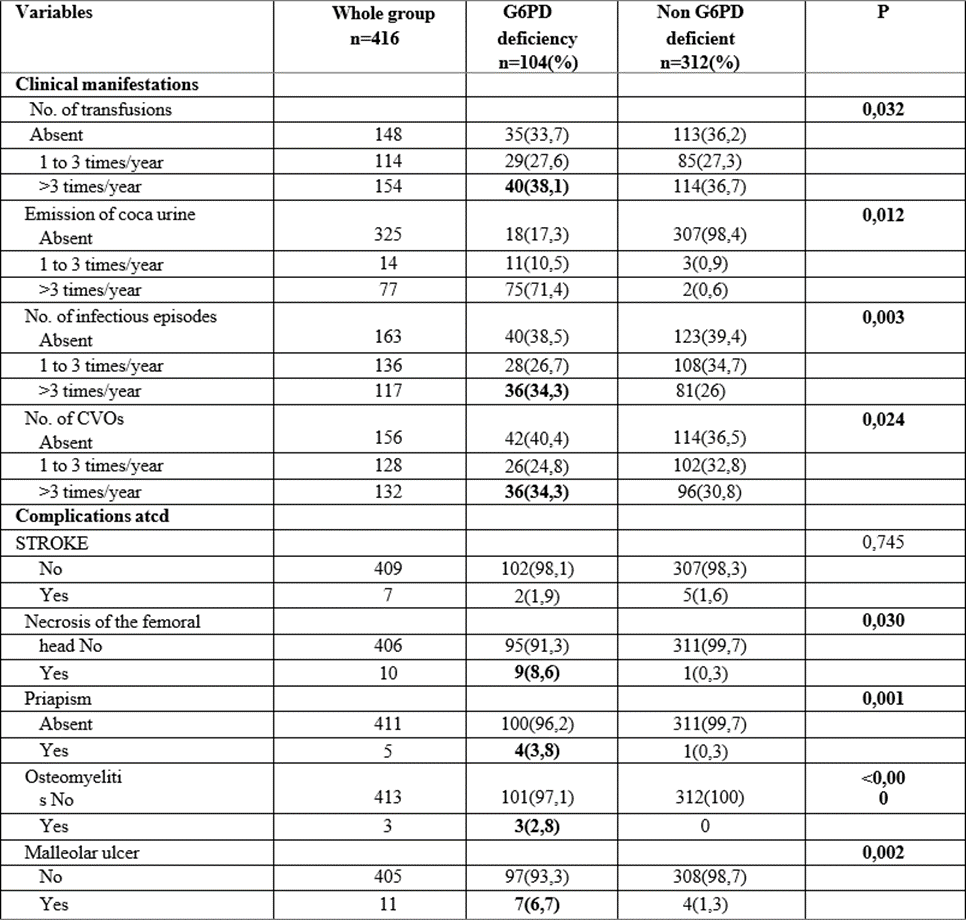
Table 2: Clinical information of G6PD-deficient and non-deficient children with sickle cell disease.
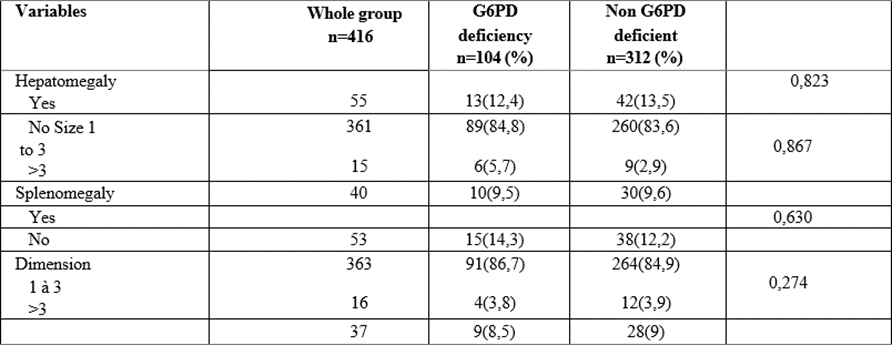
Table 3: Physical signs of G6PD-deficient and non-deficient childrenwith sickle cell disease.
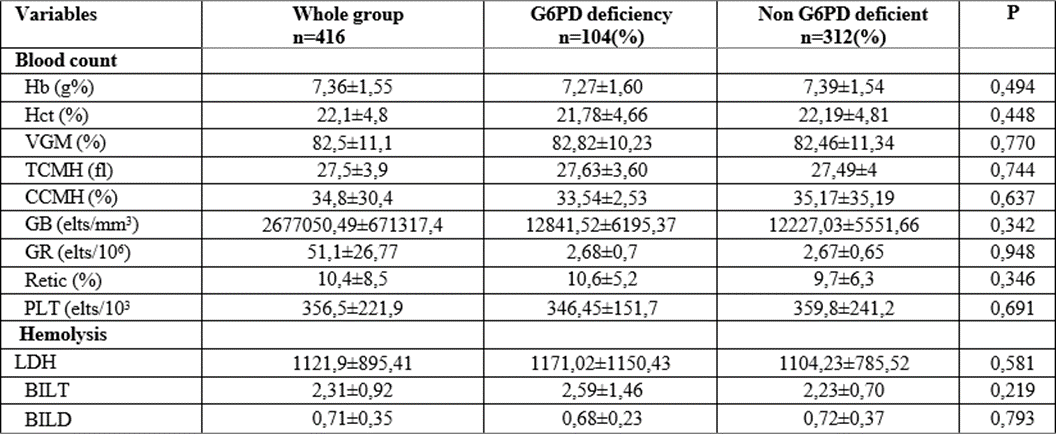
Table 4: Biological data of sickle cell patients according to their G6PD deficiency status or not
The association between sickle cell disease and G6PD deficiency is very high in regions of high malaria endemicity, particularly in sub-Saharan Africa. The Democratic Republic of Congo (DRC) is no exception.
Clinically, our study showed that all morbid manifestations (the number of transfusion episode, coca- cola urine emission, infectious episode and vaso- occlusive crises) occurring in sickle cell patients were greater in the group with G6PD deficiency (p˂0.05). Previous complications (osteomyelitis, malleolar ulcer, and priapism) evaluated occurring in sickle cell patients were associated with G6PD deficiency (p˂0.05), with the exception of stroke. No statistically significant difference had been noted for physical signs between sickle cell deficient and nondeficient groups (p>0.05).
Our results agree with those of Mathula et al, in 2012 in the USA, Benkerou et al, (2013)as well as Bernaudin et al, in France in 2018, Lohit et al, in India in 2019, Mounkaila et al, in 2015 in Niger, and Bouanga et al, in 1998 in Congo, who reported that there were more transfusions in the G6PD- deficient sickle cell group, but found no significant difference in vasoocclusive crises and infectious episodes [2, 8-11, 21].
In contrast, Foluke et al. in 2019 in Nigeria, Diop et al. in 2005 in Senegal, Simpore et al. in Burkina Faso in 2007, Gbadoe et al. in Togo in 2001, and Firempong et al. in 2016 in Ghana found no significant difference in clinical parameters between G6PD-deficient and non-G6PD- deficient sickle cell disease[3-5,7,22].
We can justify these discrepancies by the fact that the sickle cell haplotype found in the DRC, which is the Bantu haplotype, is the most severe form, in contrast to the Senegal, Benin, Cameroon and Arab-Indian haplotypes; and the variant of G6PD deficiency found in Africans associated with sickle cell disease is type A which is a genetic form of less severity contrary to variants B (Mediterranean), Mahidol, Canton, Guadalajara, Beverly Hills, and Nashville which are more found in America, Europe and Asia and which are more severe forms [23, 24].
Indeed, the disorders related to G6PD deficiency and sickle cell disease are due to the oxidative effect, which is an imbalance between the production of free radical sand the body's ability to detoxify their harmful effect due to the neutralization through antioxidants. Free radicals are usually formed during normal metabolic reactions, but the body manages to detoxify them and decrease their level in the cell through the antioxidant effect. Sickle cell patients produce a fair amount of free radicals responsible for clinical manifestations and complications causing several organ damages including vaso-occlusive disorders [9, 25]. These free radicals also cause damage to membrane proteins and lipids leading to destruction of RBCs resulting in hyper hemolysis attacks and other morbid manifestations in sickle cell children [26].
There is still much controversy to this day about the impact of G6PD deficiency on sickle cell disease. This is the case of Piomelli et al, (1972), who argue that these two abnormalities seem to be mutually attenuating: sickle cell disease making the deficiency enzyme to the extent that the deficient genotype appears to be masked [6].
The discrepancies and similarities found in the literature can also be explained by various phenomena that influence the course of sickle cell disease. We mention the hereditary persistence of fetal hemoglobin, the association with alpha thalassemia and the haplotype membership; all these elements were not sought in our study[27, 28].
Referring to the biological data of the studied children, the blood count as well as the hemolysis markers showed no difference between the two groups taken in steady state (p>0.05).
These observations meet those of Diop et al. in Senegal, Firempong et al. in Ghana in 2016 [3, 7]. However, Mehdi et al, in the USA in 2010, Bernaudinet al, (2018), and Benkerou et al, (2013) in France, as well as Lohit et al, in India in 2019, Foluke et al, in Nigeria in 2019, found that G6PD-deficientsickle cell patients had lower hemoglobin levels and elevated reticulocytes [2,4,10,29].
This is justified by the fact that in our study, as in the other African studies that found similar results to ours, the biological parameters were evaluated in a steady state, our patients had been in a stable state for about three months before our visit.
It is described that in case of hemolysis, there is a decrease in the number of RBCs, which also results in a decrease in the hemoglobin level, leading to marrow activation with an increase in the number of reticulocytes, which are immature RBCs; this would justify the increased episodes of hemolysis and anemia in G6PD- deficient sickle cell patients.
Our results should not obscure the fact that the association of sickle cell disease and G6PD deficiency carries obvious potential risks. Indeed, most of the drugs used by sickle cell patients are oxidizing and therefore likely to cause hemolysis in deficient patients. It is therefore useful to screen sickle cell patients for G6PD deficiency in order to derive preventive and therapeutic measures.
Study limitations and strengths Study limitation
The single measurement of biological parameters may have led to the under or overestimation of the values obtained.
Strength of the study
Beyond its limitations, the present study has, nevertheless, some strengths. It is the very first large study, which evaluates, through 4 health districts of the city and province of Kinshasa, the frequency and the clinico-biological impact of G6PD deficiency in children with sickle cell disease in DRC. This will certainly contribute to raising awareness in the management of these patients.
The present study aimed to determine the impact of G6PD deficiency on sickle cell disease in children in Kinshasa. Thus, the study showed that G6PD deficiency is associated with acute clinical manifestations as well as complications of sickle cell patients, including: the occurrence of vaso occlusive crises, malleolar ulcer, osteomyelitis, aseptic necrosis of the femoral head, and priapism without influencing the occurrence of stroke. And it has no impact on the hematological parameters of sickle cell children in stationary phase. Hence the need to systematically search for G6PD deficiency in sickle cell patients in order to take adequate preventive measures in relation to the possible use of oxidizing drugs.
Conflict of interest: No conflicts of interest have been declared by the authors.
Authors' contributions: Ariane KETO and René NGIYULU conceived, collected, interpreted, wrote and corrected the manuscript. Gibency Mfulani and Eric Mafuta analyzed the data, read and corrected the article. Joseph BODI, Léon TSHILOLO, Loukia Aketi and Aléine BUDIONGO supervised, interpreted and corrected the article. Balthazar PHOBA and Yves MUZINGA analyzed the samples in the laboratory of the Monkole Hospital Center. All authors read and approved the final version of the article.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
