
Research Article | DOI: https://doi.org/ 10.31579/2768-0487/017
Laboratoire de Recherches et Développement des médicaments et des produits naturels, Faculte of Pharmacy, Lebanese University, Beirut, Lebanon Department of Veterinary Sciences, Faculty of Agriculture, Lebanese University, Dekwaneh, Lebanon.
*Corresponding Author: Ali Jaber, Department of Veterinary Sciences, Faculty of Agriculture, Lebanese University, Dekwaneh, Lebanon.
Citation: Z Zahreddine, A Jaber, Simon A Haidar, C Hosri, G Ibrahim et al. (2021) HPLC-DAD multi-residue method for determination of florfenicol, penicillin and tetracycline residues in raw cow milk. Journal of Clinical and Laboratory Research. 2(3) DOI: 10.31579/2768-0487/017
Copyright: © 2021 Ali Jaber. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 29 March 2021 | Accepted: 19 April 2021 | Published: 22 April 2021
Keywords: antibiotics; HPLC-DAD; raw milk; SPE
The existence of antibiotic residues in edible products constitutes a health problem to the consumers. Reversed-phase high-performance liquid chromatography with diode array detection (HPLC–DAD) was optimized and validated for the simultaneous determination of florfenicol (FF), penicillin (PE), and tetracycline (TC) residues in dairy raw milk samples. The determination of these antibiotics was carried out on HP-ODS Hypersil C18 (5μm, 125*4 mm) column at a flow rate (1mL/min) and temperature (35 ⁰C). The extraction method includes deproteinization of the milk sample followed by a solid-phase extraction (SPE) clean-up. The method was validated according to the European Commission Decision 2002/657/EC and the International Conference of Harmonization Guidelines. The recoveries for the studied antibiotics ranged from 82–111.54 % making the method suitable for performing routine analysis. The proposed method was applied for the analysis of antibiotic residues in 50 dairy raw milk samples collected from many regions in Lebanon. The results showed the occurrence of these antibiotics residues in milk collected from different Lebanese regions. The numbers indicate that 22 % of milk samples were found to be positive for FF, 42 % for PE, and 28 % for TC residues.
Antibiotics (ATBs) are vital drugs implemented in the treatment of animal bacterial infections. Their effectiveness is, however, threatened by extensive and imprudent use, not only in cattle but also in human medicine. In veterinary practice, ATBs are utilized at therapeutic levels primarily to treat diseases and to prevent infections [1]. However, they are used at sub-therapeutic levels to increase feed efficiency, promote growth and prevent diseases [2]. Thus the imprudent use of ATBs may be a cause for the presence of ATB residues in dairy products and constitutes a real risk to consumers. Since they can lead to allergic reactions in hypersensitive individuals, and they may result in drug-resistant bacteria [3]. Hence, the ATB residues’ monitoring is needed to guarantee food safety.
To ensure food safety, regulatory authorities such as Codex Alimentarius Commission (CAC) [4] and the European Union (EU) [5] have enacted strict Maximum Residue Limit (MRL), while the US Food and Drug Administration (FDA) establishes the tolerance or safe level [6]. As well as, those authorities’ have imposed strict requirements concerning the performance of analytical methods and the interpretation of the results. It should be noted that MRLs in a particular product may differ from one location to another and most developing countries have yet to develop their own MRLs [7].
β-lactam ATB, including penicillin (PE) (Figure 1) derivatives, are among the widely used ATBs in veterinary medicine precisely due to their high specificity, perfect selective toxicity, and potent killing effects [8,9]. Moreover, tetracyclines (TC) (Figure 1) are considered as the first-line drugs in food animals [10]. TC residues can cause harmful effects, such as allergic reactions, liver damage, yellowing of teeth, and gastrointestinal disturbance [11,12]. Likewise, florfenicol (FF) (Figure 1) is a broad‑spectrum antibiotic licensed for use in veterinary medicine. FF demonstrates good tissue penetration, due to its lipophilicity character, increasing its activity against bovine respiratory disease (BRD) [13]. However, this drug was also found to penetrate most body tissues including milk of lactating cattle and goats after intravenous or intramuscular administration[14].
Thus control authorities such as Codex Alimentarius have recommended the MRLs of 100 ppb, 4 ppb, and no value respectively for TC, PE, and FF in milk [4]. The absence of FF from the list is since the FDA establishes that any quantity of FF in milk is considered violative [15,16]. Despite the FF tolerance has not been established by the FDA, some studies suggest that the maximum provisional acceptable residues of FF in milk could be approximately in a range between 80 and 200 ppb [17].
Different methods, such as microbiological and chromatographic methods have been described for monitoring antibiotics in milk. Bioassay techniques are less specific and sometimes, they produce false positives [11]. Chromatographic techniques, such as thin‑layer chromatography, capillary electrophoresis and, high‑performance liquid chromatography (HPLC), have been developed for the quantitative, accurate, and reliable measurements of antibiotics in milk and animal tissues [11]. Despite the development of a wide range of multiclass methods dedicated to simultaneously analysis residues belonging to different ATB families, to the best of our knowledge there is no HPLC method allowing the analysis of FF, TC, and PE concurrently [18,19].
In Lebanon, small farmers are not subject to follow-up by the relevant ministries, and production is marketed directly to citizens without being subject to any medical checks. Moreover, studies dealing with these products are almost non-existent, firstly due to the lack of sufficient awareness by citizens [20] and secondly to the refusal of farmers to cooperate as a result of fear or for personal unknown reasons. Otherwise, some studies only dealt with milk samples from common local brands available in the Lebanese market [21] or with red meat [20]. Moreover, Lebanon is classified to be among the countries which have a high intake of milk defined as per capita milk consumption/year of >150 kg [22]. It’s worth noting that studies reported a high prevalence of resistant bacteria in the Lebanese community, especially penicillin [23].
The aim of this study was first to develop and validate a solid-phase extraction (SPE)-HPLC-diode-array-detector (DAD) method for the simultaneous separation and quantitation of three target ATB in raw cow milk. The applicability of the proposed method was demonstrated later by using it to investigate the incidence of those ATBs in raw milk samples collected from dairy farms in many regions in Lebanon. The samples have been analyzed for 3 ATBs compounds FF, TC, and PE.
Sample collection
A total of 50 raw cows' milk samples, from Lebanese dairy farms, were randomly collected throughout the year in sterile sample containers. Samples were labeled and stored at -20 oC for further analysis.
Chemical and Reagents
HPLC gradient grade acetonitrile and methanol were purchased from VWR chemicals. Oxalic acid and disodium hydrogen orthophosphate anhydrous were obtained from Analar. Citric acid anhydrous was purchased from HIMEDIA Laboratories. Disodium ethylenediaminetetraacetate (EDTA), Penicillin G potassium salt, and Formic acid (FA) were purchased from Sigma-Aldrich. Tetracycline, Florfenicol was generously provided by Pharmadex S.a.l. (medicine factory, Beirut, Lebanon). Ultra-pure water (TKA, Micromed, Germany) was used for the preparation of all aqueous solutions. The solid-phase extraction procedures were carried out using Waters SupelTM-Select HLB cartridge (200 mg, 6 mL) were provided from Sigma-Aldrich.
Apparatus
All measurements were accomplished using an HP 1100 Series LC system (Hewlett Packard, Palo Alto, CA, USA) equipped with a quaternary pump, a vacuum degasser, a column compartment, an auto sample, and a diode-array detector, and controlled by the HP Chemstation chromatography software. The analytical column was ODS hypersil C18, 5 μm (125 x 4mm) (from Hewlett Packard, Palo Alto, CA, USA). Other equipments such as pH meter CG 820 (SCHOTT GERATE, made in West Germany), electronic weighing balance (RADWAG Wagi Electronic, Poland), Spectrafuge 6C compact centrifuge (Edtexison, NJ USA), Ultrasonic cleaner (BRANSON 200, made in Taiwan) and vortex made by Daihan Scientific Co. (Korea) are also used in this study.
Preparation of Standard Solutions
To obtain a final concentration of 1mg/mL, a stock standard solution of FF, TC, and penicillin was prepared by dissolving 1 mg of the compound in 1 mL of acetonitrile (ACN), methanol (MeOH), water/ACN (1/1) respectively. The solutions were stored at + 4 °C until further use. Working solutions were prepared daily by appropriate dilution of aliquots of the standard stock solutions in ultra-pure water. The working solutions were used for sample spiking for the preparation of calibration curves of 6 different concentrations.
Extraction and clean-up procedure
About 10 mL of milk sample was taken in a 50 mL centrifuge tube. Add to it 10 mL of 0.1M EDTA-McIlvaine buffer (pH 4.0) (prepared as described by Cinquina et al.[24]) followed by vigorous shaking for 5 min. The sample was then centrifuged at 6000 rpm for 10 min. The supernatant was collected and filtered through a Whatman filter paper 0.45 μm (Whatman, Maidstone, UK) to remove any remaining milk flakes. Clean up of the extract was done by using the SPE method. The filtrate was loaded on a Supel Select HLB (Hydrophilic-Lipophilic Balance) cartridge preconditioned with 3 mL of methanol followed by 2 mL of ultra-pure water under pressure. The cartridge containing the sample was washed with 2 mL of water and then antibiotics were eluted with 1.5 mL of MeOH. The obtained elute was filtered through a 0.45 μm syringe filter and stored in vials for further analysis.
Chromatographic conditions
The LC gradient elution was performed using a mobile phase consisting of solvent A: water (containing 0.1 % FA), solvent B: oxalic acid (0.05 M and pH = 2.6)/ ACN/MeOH (60:30:10), solvent C: 25 % ACN in water (containing 0.1% FA) and solvent D: ACN/MeOH (2:1) (containing 0.1% FA). The mobile phase was mixed and sonicated for 5 min and then vacuum filtered through a 0.45 µm nylon filter.
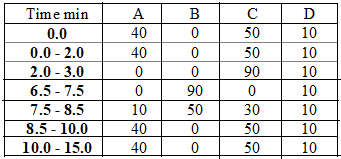
Chromatographic separation of the analytes was achieved on ODS hypersil C18 column. An elution gradient was chosen that allowed complete analysis in less than fifteen minutes; the characteristics are reported in Table 1 below. The flow rate was adjusted at 1 mL/min and the column thermostat was set at 35 oC. The injection volume was 25 μL and the final run time of the method was 15 min. Detection wavelengths were set at 224 nm for FF, 210 nm for PE, and 350 nm for TC. While data analysis was performed utilizing the Hewlett-Packard ChemStation software.
In-House Validation of the Analytical Method
The characteristics and the procedures used for validation were performed following the recommendations from the Commission Decision 2002/657/CE of the EU[25], for the parameters of retention time, linearity, recovery, and precision. The LOD and LOQ were calculated according to the guideline of the International Conference of Harmonization (ICH) guidelines [26]. The performance criteria; linearity, sensitivity, selectivity, intra-assay and inter-assay precision, accuracy, the limit of detection (LOD), and limit of quantitation (LOQ) were determined.
The linearity response was examined by triplicate analysis of milk samples fortified with FF, PE, or TC at seven fortification levels ranging from 0.004 to 5 ppm. The standard calibration curves were generated for each analyte by plotting concentrations against the peak height ratio of the analyte.
The sensitivity of the method, i.e. the change in response on a measuring instrument divided by the corresponding change in stimulus, was represented by the slope of the calibration curve [27].
The selectivity of the method was investigated by analysis of ten different blank milk samples to determine any interfering peaks from endogenous compounds.
LOD and LOQ established for this method were calculated from the standard deviation (σ) of y-intercepts of regression analysis and the calibration curve slope (m), according to equations 1 and 2 respectively [26].
LOD=3.3σm (eq. 1)
LOQ=10σm (eq. 2)
The precision of the method consists of intra-assay precision and inter-assay precision expressed as a percentage of relative standard deviation (% RSD) of peak height measurements. The intra-assay precision was determined by spiking six blank milk samples at a single concentration level of 5 μg.mL-1 and evaluation was done through the results obtained with the method operating over 1 day under the same conditions. The inter-assay precision was determined at three fortification levels, and the analyses were carried out over three consecutive days.
Since no certified reference material was available, the accuracy of the method expressed as % recovery was determined by triplicate analysis of spiked milk samples at three fortification levels. The recoveries were calculated by comparing the mean value of the measured concentration to the mean value of the spiked concentration. The below equation (eq.3) was used to calculate the apparent recovery.
%Recovery = obtained mean valueexpected value . 100 (eq.3)

Optimization of Chromatographic Separation
The development of a simultaneous SPE-HPLC-DAD method to isolate FF, PE, and TC, and separate them from matrices is easier said than done. Since the three targeted ATBs belong to different families, thus they have distinct physic-chemical properties. Optimization of several chromatographic parameters was performed to obtain the best chromatographic conditions. So, a series of experiments concerning modification of mobile phase, stationary phase, column temperature, and absorption spectrum (wavelengths) were evaluated.
First, several mobile phase compositions and gradient profiles were investigated to improve peak separation and to obtain the shortest run time. Our first choice was the elution gradient reported by Karami-Osboo et al. (2016) to FF analysis with a mobile phase consisted of water/acetonitrile 75:25 (vlv) [28]. When used for blank milk samples, the later mobile phase leads to an interfering peak in the retention time-window of FF. To improve the FF elution selectivity we had resorted to adding 0.1% FA to this mobile phase, increasing the retention time of FF. According to literature studies, β-lactam ATB has been separated using acidic mobile phases [29,30]. Also, the problem of tailing peak, due to metal impurities or residual silanols when using reversed‑phase column, can be overcome by adding acid to mobile phase [31]. However, these conditions allowed the elution of PE and FF only. To optimize the TC elution and separation, other experiments were done. The addition of oxalic acid (0.05 M) and 0.1% acetic acid was tried since the buffer type has a critical impact on the protonation of compounds. As result, increasing the percentage of oxalate buffer in the mobile phase provides the best results. Similar findings concerning the effect of oxalic acid were observed [32], and the given explanation was related to the tendency of oxalate to block the residual silanols group on the column. From another side, experiments were done to select the best organic modifier of the mobile phase, and conclusions were drawn that methanol provides superior separation and sensitivity to the antibiotics in comparison to acetonitrile only. Further, it was reported that when the MeOH concentration increased in the mobile phase the elution time becomes short [33].
A column choice is another important step in the development of multi-residue HPLC methods. Three different LC columns (HP ODS Hypersil C18 (5μm, 200*4.6 mm), HP ODS Hypersil C18 (5μm, 125*4 mm), and Nucleosil C18 (125*4mm) were compared and used during the initial elaboration of the method. The ODS Hypersil C18 (5μm, 125*4 mm) column indicated a rapid and better chromatographic separation of the analytes and gave the best sensitivity with the chosen compositions of the mobile phase. This column was used for all further method development and validation experiments.
Other conditions namely the column temperature and maximum absorption wavelength were optimized to incorporate FF, PE, and TC. Thereby, once adjusting the column temperature to 35 °C, excellent separation for these 3 antibiotics was achieved on the ODS Hypersil C18 column using a photodiode array detector. The detection wavelengths used to attain maximum sensitivity were 224 nm for FF, 210 nm for PE, and 350 nm for TC. Finally, the overall optimized conditions have been applied using a mixed standard solution and showed a good selectivity in chromatograms of raw milk samples for three ATB. Very low variation was observed in the retention times, with RSD values not exceeding 1.65%. Thus, the optimized method resulted in an effective separation of the three antibiotics belonging to three different groups in a single run with adequate resolution.
Pre-cleaning of milk samples
Sample preparation is a critical technical stage of the development of multi-detection and multi‑class methods, as it is important to ensure good purification providing maximum removal of interferences and enhance the recovery of the analytes.
As mentioned earlier, the development of a multi-class method is a difficult task due to different physicochemical properties between classes. This requires pre-treatment as well as clean-up of samples to eliminate specific interferences from the milk matrix facilitating analytical determination of each analyte with adequate resolution. The four main steps evaluated were precipitation of milk proteins, extraction of ATB from milk matrix, cleanup using solid-phase cartridges, and concentration of eluting. For extraction studies, the known blank milk samples fortified with ATB standard solutions (1 µg.ml-1) were employed.
In order to evaluate the precipitations’ step of milk proteins, different solutions such as trichloroacetic acid, acetonitrile, and EDTA Mcllvaine buffer have been evaluated. The best results were obtained from sodium EDTA-Mcllvaine buffer that resulted in simultaneous extraction of all targeted antibiotics from milk with good recoveries. The sodium EDTA-Mcllvaine [34] buffer has been used by many researchers for the extraction of antibiotic residues from milk [35]. The extracts that had undergone the protein precipitation were subsequently cleaned up using Speed SPE C18 solid‑phase extraction cartridges.
For the selection of SPE sorbent, the extraction efficiency of two SPE columns (Waters Sep-Pak Vac and Supel Select HLB) were tested and they give roughly the same recovery values. Supel Select HLB cartridge was used for the accomplishment of the work. After loading, 2 mL of ultra-pure water was used for the washing step. The final elution step was carried out using 1.5 mL of methanol, then filtered through 0.45 µm syringe filters and transferred into vials for HPLC analysis.
Validation of Method
The optimized HPLC-DAD method was validated for the determination of the three ATBs (FF, PE, and TC) and results are presented in Table 2.

The linearity and the sensitivity were established by the calibration curves by plotting the peak height against concentrations of each analyte ranging from 0.004 to 5 ppm. Each concentration level was injected three times (n = 3). Good linearity was obtained for all ATB and highly correlated with the amounts injected, the correlation coefficients (r2) ranged from 0.997 to 0.999.
The food matrix imposes a lot of interferences and the ability of the method to quantify a particular analyte despite all interferences is measured by the selectivity [36]. The application of the method to different blank milk samples demonstrated that no potential interferences from the matrix were detected at the retention time-windows of three analytes. Therefore, the optimized method presents adequate selectivity for the determination of the studied ATB. Representative chromatograms of blank samples and fortified samples are shown in Figure 2.

To fulfill the requirements of the legislated MRLs, the limit of detection (LOD) and limit of quantification (LOQ) were calculated using the aforementioned (eq. 1) and (eq. 2) respectively. As presented in Table 2, the LOD for the studied antibiotics ranged from 6 to 12.4 ppb, while the LOQ ranged from 18 to 45 ppb.
To assess the precision of the method, intra-assay and inter-assay (expressed as % RSD) were checked. Repeatability of the method was tested by six replicate injections of spiked milk. Intra- and inter-day variations of retention times and concentrations, expressed in RSD %, are summarized in Table 2. % RSD values for antibiotic concentration ranged from 0.77 and 1.68 %. According to the European Commission Decision 2002/657/EC [37], the intra-assay precision and inter-assay precision should be lower than 15 % and 23 %, respectively, and the observed values are in agreement with the EU guidelines.
Accuracy was established based on ATB recovery, which was determined by triplicate analysis of spiked milk samples at three fortification levels (low, medium, and high). The recovery for studied antibiotics ranged from 82 to 111.54 %, and the results are reported in Table 2. The recovery values are under the EU guidelines, which established a range of 80–120 % for these concentration levels [37]. Consequently, it can be noticed that the proposed method is accurate and precise enough for the determination of ATB residues in raw milk.
Milk Samples Analysis
To verify the performance of the proposed method, 50 samples of pooled raw milk procured from randomly selected dairy farms located in Lebanon, were analyzed for the presence of targeted ATB. The obtained HPLC results (Figure 3) indicate that 22 % of milk samples were found to be positive for FF, 42 % for PE, and 28 % for TC residues.
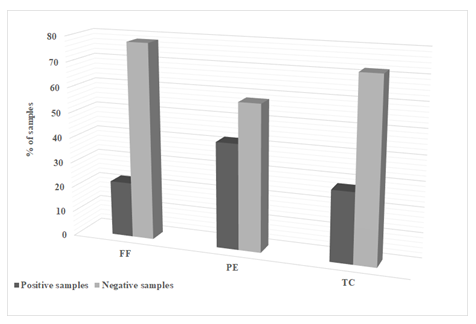
Results were categorized according to the obtained concentrations and with levels above the established MRLs values [37]. Positive samples could be probably due to the misuse of antibiotics in food animal production in the Lebanese farms, together with the violation of the withdrawal period regulation.
Our results showed that PE was the most prevalent ATB in milk samples. The occurrence of β-lactam ATB can be attributed to their frequent use in the treatment of mastitis and other systemic diseases [38]. The obtained result, (42 % of samples positive for PE) was in correlation with studies conducted in Italy [39] and Turkey [40], which have shown the presence of β-lactams residues at concentrations higher than the MRLs. In addition to the above studies, results are enhanced by another study conducted in Northern Italy, as most of the ATB residues found belonged to the group of β-lactams [41].
In Kuwait, Alomirah et al. [42] collected samples from approximately 1000 locally produced and imported milk and dairy product at different seasonal periods from different farms and retail outlets and screened for the presence of four antimicrobial residues (beta-lactams, tetracyclines, sulfonamides, and chloramphenicol). Results indicated that 29.1% of the analyzed local fresh milk samples were above the MRL for tested residues with TC as predominant residue [42]. Similar results were found in a study conducted in Macedonia [43]. Controversy, Bilandzic et al. [44] performed a study on 119 raw milk samples (in Croatia) and the measured mean value for TC was 2.83 μg/l where none of the samples analyzed showed the presence of veterinary drug residues above the MRL. Despite the few studies examining the presence of FF residues in raw milk, we can find two interesting studies in the last five years. A study done by Karami et al. [28] on 15 real milk samples analyzed for florfenicol and chloramphenicol and the results were only one sample was contaminated with florfenicol and contamination to chloramphenicol was not detected. A similar result was obtained recently by Wang et al. [45]. Following our results, the prevalence of FF residue has a limited presence in raw milk.
On the other hand, the prevalence of multi-class residues in one sample might be due to several reasons, such as the use of medicated feed or simultaneous use of different types of antibiotics intravenously/systemically or locally at the udder [46].
Based on this work, urgent stricter rules turn out to be a must on the use of ATB in animal husbandry if the authorities have the firm intention to decrease the level of residues in milk samples. This study emphasized the importance of education and awareness programs concerning the respect of the withdrawal periods of antimicrobials as well as stress the importance of the supervisory role of institutions concerned with food safety.
To the best of our knowledge, this is the first study in which a fast and reliable method has been developed and validated for simultaneous detection and quantification of FF, PE, and TC in raw milk. The developed method provided good performance and satisfactory recovery, thus results showed the applicability for routine analysis of raw cow milk. Then, the validated method had served to detect and quantify FF, PE, and TC residues in a reduced number of samples from Lebanese farms. The overall results showed that the prevalence of these antibiotics residues in milk was high in terms of contamination among the collected samples due to a lack of surveillance and monitoring system from the concerned authorities. Our findings highlight the need for antibiotic monitoring among small farms and this study would serve as baseline data on the prevalence of antibiotics residue in raw milk.
The authors gratefully acknowledge the financial support from Lebanese University (Faculty of Pharmacy).
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
