
Case Report | DOI: https://doi.org/10.31579/2641-0419/360
1Department of Cardiology, Sree Chitra Tirunal Institute for Medical Science and Technology.
2Department of Medicine, North Bengal Medical College, Siliguri, WB, India.
*Corresponding Author: Sudipta Mondal, Department of Cardiology, Sree Chitra Tirunal Institute for Medical Science and Technology.
Citation: Sudipta Mondal, Dipanjan Bandyopadhyay, (2024), Holt-Oram Syndrome – A Case Report and a Short Review, J. Clinical Cardiology and Cardiovascular Interventions, 7(3); DOI:10.31579/2641-0419/360
Copyright: © 2024, Sudipta Mondal. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 21 March 2024 | Accepted: 29 March 2024 | Published: 11 April 2024
Keywords: holt-oram syndrome; atrial septal defect; asd; absent radius; case report
Holt-Oram syndrome also known as the heart-hand syndrome type 1, is an autosomal dominant rare congenital disorder, manifested as a conglomeration of various skeletal and cardiac abnormalities. The list of differential diagnoses of such abnormalities is extensive but careful examination can point towards a specific diagnosis even without genetic testing in a resource-limited country. We present a sporadic case of Holt-Oram syndrome with OS-ASD highlighting the diagnostic problem without genetic testing and discussing differentiating salient features of most of the syndromic associations.
Holt-Oram syndrome, a rare congenital disorder with a conglomeration of various defects especially skeletal and cardiac abnormalities, is an autosomal dominant disorder that was first described by Holt and Oram in 1960 in family members across four generations.1 Differentiation of closely related phenotypic disorders is possible with good clinical examination even in the absence of genetic testing in a resource-limited country, as cardiac involvement is not the same for all. We present a case of Holt-Oram syndrome with cardiac involvement and a brief review of differentiating close mimickers (Table 1).
| Holt-Oram syndrome | Thrombocytopenia absent radius syndrome | Phocomelia | Ellis-van Creveld syndrome | |
| Synonyms | Heart-hand syndrome | TAR syndrome | Thalidomide syndrome | Chondroectodermal dysplasia Mesoectodermal dysplasia |
| Incidence | 1 in 100,000 (M=F) | 0.5-1 in 100,000 (M=F) | 0.62 in 100,000 (M=F) | 0.5-1.6 in 100,000 (M=F) |
| Inheritance pattern | AD | AR | Undetermined > Thalidomide toxicity | AR |
| Gene | TBX5 | RBM8A | - | EVC2 |
| Chromosome | 12q24.1 | 1q21.1 | - | 4p16.2 |
| Classical skeletal involvement | Thumb (+/- or hypoplasia) Absent radius (+/- or hypoplasia) | Bilateral absence of the radii with the presence of both thumbs (100%) Lower limb defects (50%) | Absence or hypoplasia of arm/forearm with near normal hands/ feet One limb (56%) Two limbs (40%) UL>>LL Left>Right | Short-limb dwarfism Partial cleft lip Defect in alveolar ridge, Hypodontia Centrifugal shortening of limbs
|
| Predominant cardiac involvement | OS-ASD > VSD Conduction abnormalities | OS-ASD > VSD >> TOF (17%)
| Uncommon (TOF) | OP- ASD Single atrium |
| Mental retardation | - | - | - | ++/- |
| Thrombocytopenia | - | + (usually <50> | - | - |
| Cow’s milk allergy | - | + | - | - |
| Gastrointestinal/ Genitourinary anomalies | +/- | +/- (~25%) | +/- | +/- |
TAR - Thrombocytopenia absent radius; AD – autosomal dominant; AR – autosomal recessive; TBX5 – T-Box Transcription Factor 5; RBM8A - RNA-binding motif protein 8A; EVC2 - Ellis van Creveld syndrome 2; UL – upper limb; LL – lower limb; OS-ASD – ostium secundum atrial septal defect; VSD – ventricular septal defect; TOF – tetralogy of Fallot; OP-ASD – ostium primum atrial septal defect.
Case: A 45-year-old man presented with NYHA functional class II dyspnoea for 6 months and was referred. He denied any history of recurrent lower respiratory tract infection or cyanosis. There was no significant family history. Clinical examination revealed no facial dysmorphism, normal hairline, no chest cage abnormalities, and no significant dental or eye abnormalities. Upper limb examination showed asymmetric limb shortening (right > left), shorter humerus on the left side, absent radius bilaterally and short ulna bilaterally (Figure 1A, B, C). There were symbrachydactyly and syndactyly. The thumb was absent bilaterally. Lower limbs were normal.
Cardiovascular examination was suggestive of pre-tricuspid septal defect with a significant left to right shunt. Other system examinations did not reveal any abnormalities. The electrocardiogram showed an incomplete right bundle branch block with a normal PR interval with an axis of -30 degrees. Chest X-ray showed increased pulmonary blood flow and mild cardiomegaly. The complete blood count was normal. The echocardiogram revealed normal left ventricular systolic function, dilated right ventricle, right ventricular systolic pressure of 45mmHg, and 30mm ostium secundum atrial septal defect (OS-ASD) with a left to right shunt (Figure 1D). He is awaiting a device closure.
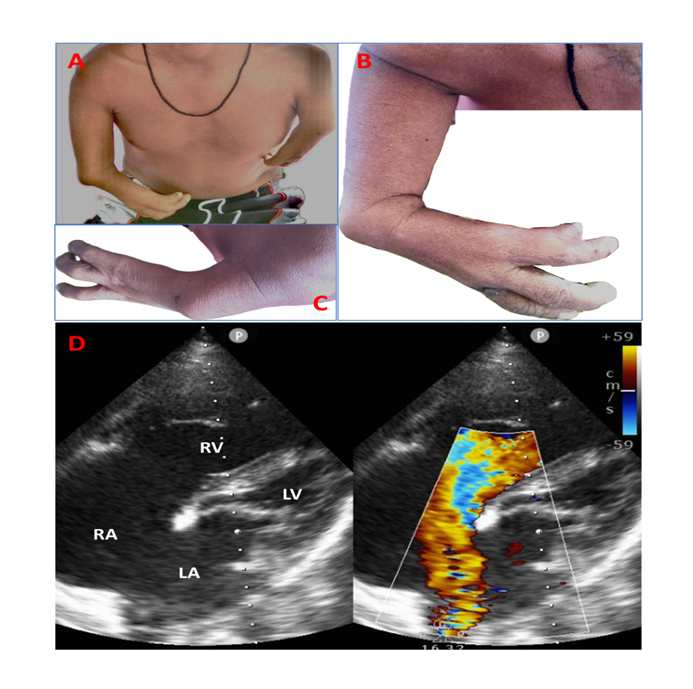
A, B, C: Clinical image of patient 1 showing asymmetric limb shortening (right > left), shorter arm and forearm on the left side. There is symbrachydactyly and syndactyly. There are four and three fingers on the right and the left side, respectively. The thumb is absent bilaterally; D. 2D echocardiogram in apical 4-chamber view showing large ostium secundum atrial septal defect with left to right shunt with dilated right atrium and ventricle.
Holt-Oram syndrome also known as the heart-hand syndrome type 1, is an autosomal dominant disorder that is characterised by upper limb skeletal defects in conjunction with congenital heart lesions. This is caused by heterozygous mutation in the TBX5 gene (encoding T-box5 transcription
factor) on chromosome 12q24.1, which is responsible for cardiac septation and development of limbs. Nearly 58% of the clinically suspected Holt-Oram syndrome carry this mutation.2 60% of the cases are familial, the rest being sporadic.3 It is reported to have 75% penetrance for cardiac defects. Wide phenotypic variability and de novo mutation make the prediction of malformation difficult.4 The incidence of Holt-Oram syndrome is around 0.001% with no sexual predilection.
The classical manifestations are upper limb malformation, congenital heart malformation, and conduction system defects. Upper-limb skeletal abnormalities can be unilateral or bilateral (96%), symmetric or asymmetric (91%).2 When unilateral, mostly it affects the left side. Fingerisation of the thumb or triphalangeal thumb is one of the characteristic features of this syndrome. Absence or hypoplasia of thumb/ radius, fusion or underdeveloped carpal bones are the other manifestations.
Cardiac defects are seen in 75% of the patients, predominantly OS-ASD followed by ventricular septal defects and other rarely reported anomalies like a bicuspid aortic valve, left ventricular non-compaction, patent ductus arteriosus, coarctation of the aorta, pulmonary stenosis, left superior caval vein.2,5,6 Vanlerberg et al reported 78 patients of TBX5 mutation, of which 91% had congenital heart defects, atrial septal defect constituting around 62% followed by ventricular septal defect (35%) and atrioventricular septal defect (5%).2 Complex cyanotic congenital heart defects (1.3%) and conduction abnormalities were less common.2 Multisystem involvement is seldom reported.7 A large prospective study including 1653 patients with upper limb malformations (clinically diagnosed as Heart-hand syndrome) by Yin et al showed that 11 % had echocardiographic and 1% had electrocardiographic abnormalities, the commonest being the atrial septal defect (38%) followed by tricuspid regurgitation (37%), ventricular septal defect (6%).8 Conduction abnormalities were present only in 2.5%.8 Management is directed to specific malformations and genetic counselling is recommended in all patients with Holt-Oram syndrome. Two variants of Holt-Oram syndrome have been described in the literature.
Heart-hand syndrome type 2: It is described only in two families to date, characterized by upper limb malformations (hypoplastic deltoids, brachytelephalangy type D, mild shortening of the fourth and fifth metacarpals in some individuals, and cardiac arrhythmias.9,10
Heart-hand syndrome type 3: It is described in three members of a Spanish family to date, which is characterized by a cardiac conduction defect (sick sinus, bundle-branch block) and brachydactyly, affecting principally the middle phalanges in conjunction with an extra ossicle on the proximal phalanx of both index fingers.
Cardinal features of syndromes associated with upper limb skeletal deformity and cardiac defecthave been depicted in Table 2.
| Synonyms | Gene/Locus | Inheritance pattern | Cardiac involvement | Radial defects | Characteristic manifestations | |
| Holt-Oram syndrome2,8 | Heart-hand syndrome | TBX5 12q24.1 | AD | 90% | 100% | Thumb (+/- or hypoplasia) Absent radius (+/- or hypoplasia) |
| Thrombocytopenia absent radius syndrome11 | TAR syndrome | RBM8A 1q21.1 | AR | 15-30% | 100% | Bilateral absence of the radii with the presence of both thumbs (100%) Lower limb defects (50%) Thrombocytopenia (96%) |
| Phocomelia12 | Thalidomide syndrome | - | Sporadic | - | 100% | Absence or hypoplasia of arm/forearm with near normal hands/ feet One limb (56%) Two limbs (40%) UL>>LL Left>Right |
| Ellis-van Creveld syndrome13,14 | Chondroectodermal dysplasia
| EVC 2 4p16.2 | AR | 50-60% | 93% | Short-limb dwarfism Partial cleft lip Defect in the alveolar ridge, Hypodontia Centrifugal shortening of limbs |
| Okihiro syndrome15 | Duane syndrome Acro-renal-ocular syndrome (AROS) Duane-radial ray syndrome (DRRS) | SALL4 20q13.2 | AD | 23% | 80% | Upper limb anomalies, ocular anomalies, renal anomalies |
| Fanconi pancytopenia16 | - | Fanconi anaemia core complex gene | AR, XL | 6% | 35% | Pancytopenia, growth retardation, microcephaly |
| VACTERL association17 | - | - | Sporadic | 40-80% | 40-50% | Vertebral defects, anal atresia/ stenosis, cardiac defects, tracheoesophageal fistula, renal anomaly, limb defects |
| Townes-Brockes syndrome18 | - | SALL1 16q12.1 | AD | 9-40% | 50-90% | Anal atresia, ears malformations, sensorineural deafness, triphalangeal thumbs, renal anomaly |
| Nager syndrome19 | Acrofacial dysostoses 1 Split hand deformity -mandibulofacial dysostosis | SF3B4 1q21.2 | AD | 15% | 100% | Mandibulofacial dysostosis, deafness, absent radii/ thumbs |
| Roberts syndrome20 | - | ESCO2 8p21.1 | AR | 26% | 100% | Growth retardation, craniofacial anomaly, limb reduction defects |
| Valproate embryopathy21 | - | - | Sporadic | 14-26% | 30% | Neural tube defects, vertebral anomaly, cleft palate |
TBX5 – T-Box Transcription Factor 5; RBM8A - RNA-binding motif protein 8A; EVC2 - Ellis van Creveld syndrome 2; SALL4 - Spalt Like Transcription Factor 4; SF3B4 - splicing factor 3b subunit 4; ESCO2 - establishment of sister chromatid cohesion N-acetyltransferase 2; TAR - Thrombocytopenia absent radius; AD – autosomal dominant; AR – autosomal recessive; XL – X-linked; UL – upper limb; LL – lower limb
Table 2: Cardinal features of syndromes associated with upper limb skeletal deformity and cardiac defect
Skeletal deformities and cardiac malformations often go hand-in-hand. Proper clinical evaluation may indicate a specific syndrome that can predict cardiac involvement in syndromic patients. We present a sporadic case of Holt-Oram syndrome with OS-ASD highlighting the diagnostic problem in the absence of genetic testing.
Obtained as per COPE guidelines.
The authors declare no conflicts of interest.
All data are incorporated into the article and its online supplementary material.
Nil
None
SM (Conceptualization: Lead; Formal analysis: Lead; Writing – original draft: Lead; Writing – review & editing: Lead), DB (Conceptualization: Lead; Formal analysis: Lead; Writing – original draft: Lead; Writing – review & editing: Lead).
None
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
