
Review Article | DOI: https://doi.org/10.31579/2690-4861/178
Advisor in Pediatrics and Pediatric Psychiatry Children Teaching Hospital of Baghdad Medical City Head, Iraq Headquarter of Copernicus Scientists International Panel Baghdad, Iraq.
*Corresponding Author: Aamir Jalal Al Mosawi, Advisor in Pediatrics and Pediatric Psychiatry Children Teaching Hospital of Baghdad Medical City Head, Iraq Headquarter of Copernicus Scientists International Panel Baghdad, Iraq.
Citation: A J A Mosawi. (2022). Goldberg Shprintzen syndrome: The novel association with congenital unilateral anorchia (monorchism). International Journal of Clinical Case Reports and Reviews. 10(1); DOI:10.31579/2690-4861/178
Copyright: © 2022 Aamir Jalal Al Mosawi, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 22 September 2021 | Accepted: 27 December 2021 | Published: 03 January 2022
Keywords: goldberg-Shprintzen syndrome; absent testis (monorchism)
Background: Goldberg Shprintzen syndrome is a very rare autosomal recessive mental-growth retardation syndrome associated with characteristic facial dysmorphism, Hirschsprung disease, and a variety of neurological abnormalities, and abnormalities on brain imaging studies. However, the association of the syndrome with congenital unilateral absence of the testis (monorchism) has not been reported before. We have previously reported the thirty fourth and thirty fifth cases of the syndrome which occurred in Iraqi brothers, and described a novel therapeutic approach which was used to treat the younger brother. The aim of this paper is to report the novel association of Goldberg Shprintzen syndrome with congenital right monorchism.
Patients and methods: T.A.S, the younger of two brothers with Goldberg Shprintzen syndrome was first seen at the age of four years and 10 months at the pediatric neuro-psychiatric clinic on the 29th of August, 2019. He had spastic right hemiparesis and was unable to walk alone, and was not saying any word and had characteristic facial features including hypertelorism, narrow palpebral fissures, open mouth, and laterally lifted ear. He also had neonatal intestinal obstruction which was attributed to Hirschsprung disease, and was treated surgically with resection and colostomy. The boy was treated successfully with novel therapeutic approach and experienced improvement in cognitive abilities, speech, and motor function, and after treatment was able to walk alone.
Results: During July, 2021, the family reminded us that the child had single testis in the scrotum, and during early infancy an MRI study failed to find any second testis anywhere. An ultrasound was performed and showed normal left testis. However, the right testis could not found in the right hemi-scrotal sac nor with the right inguinal canal or within the abdomen. Thus, the ultrasound confirmed the earlier MRI findings which suggested congenital absence of the right testis (monorchism).
Conclusion: This paper reported the novel association of Goldberg Shprintzen syndrome with monorchism, and this case represented the third case of congenital syndromic monorchism in the world.
Goldberg Shprintzen syndrome is a very rare autosomal recessive mental-growth retardation syndrome associated with characteristic facial dysmorphism, Hirschsprung disease, and a variety of neurological abnormalities, and abnormalities on brain imaging studies. However, the association of the syndrome with congenital unilateral absence of the testis (monorchism) has not been reported before [1, 2, 3].
We have previously reported the thirty fourth and thirty fifth cases of the syndrome which occurred in Iraqi brothers, and described a novel therapeutic approach which was used to treat the younger brother [2, 3].
The aim of this paper is to report the novel association of Goldberg Shprintzen syndrome with congenital right monorchism.
T.A.S, the younger of two Iraqi brothers with Goldberg Shprintzen syndrome was first seen at the age of four years and 10 months at the pediatric neuro-psychiatric clinic on the 29th of August, 2019. He had spastic right hemiparesis and was unable to walk alone, and was not saying any word, and had characteristic facial features including hypertelorism, narrow palpebral fissures, open mouth, and laterally lifted ear (Figure-1A). He also had submucous cleft palate. The boy also had impaired fine motor skills and was unable to neither feed self with spoon nor drink with a cup appropriately.

There was no clear history of asphyxia at birth nor CNS injury or infection during infancy that can be blamed for the spastic hemiplegia. However, the boy had neonatal intestinal obstruction which was attributed to Hirschsprung disease, and was treated surgically with resection and colostomy at about thirty days of age.
Brain MRI showed some atrophic changes at the left parietal region. The boy was treated successfully with novel therapeutic approach and experienced improvement in cognitive abilities, speech, and motor function, and was able to walk alone (Figure-1B).

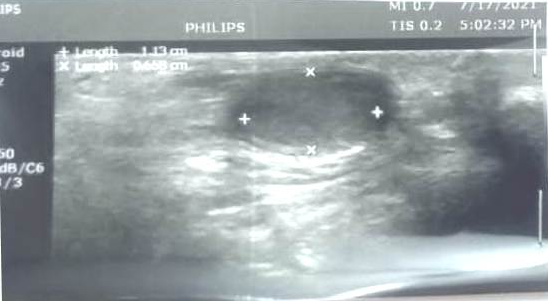
During July, 2021, the family reminded us that the boy had single testis in the scrotum, and during early infancy an MRI study failed to find any second testis anywhere. An ultrasound was performed and showed normal left testis in the scrotum (Figure-2) with a dimensions of 11 X 7 mm. However, the right testis could not found in the right hemi-scrotal sac nor with the right inguinal canal or within the abdomen. Thus, the ultrasound confirmed the earlier MRI findings which suggested congenital absence of the right testis (monorchism).
In 1981, Goldberg and Shprintzen described siblings with mental and growth retardation, characteristic facial dysmorphism, short-segment Hirschsprung disease, and cleft palate [1]. We have previously reported the thirty fourth and thirty fifth cases of the syndrome which occurred in Iraqi brothers [2], and we described a novel therapeutic approach which was used to treat the younger brother [3]. The association of the syndrome with congenital absence of the testis (monorchism) has not been reported before [1, 2, 3].
Unilateral anorchia (monorchism), congenital absence of one testis is an extremely rare condition, and the syndromic form was reported only in two patients previously [4-14]. The condition was known as early as the late 1950s [4], and few cases were reported during the 1960s and 1970s [5-9] including eight cases reported by TIBBS (1961) [6].
In 1984, Hamidinia and colleagues reported one case of unilateral anorchia [10], while Schindler et al (1987) from Switzerland reported 512 boys who had an empty scrotum. 495 (96.7%) of them had cryptorchidism, 4 of them had ectopia and 13 patients unilateral anorchia [11].
Saito and Kumamoto (1989) from Japan reported seven cases of congenital monorchism who had normal number of spermatogonia per seminiferous tubule [12].
The first case of syndromic congenital monorchism was most probably reported by Cremades Mira et al (1991) from Spain who reported a neonate with Prune Belly syndrome who had urethral obstruction, unilateral anorchia and hyaline membrane disease [13].
The second case of congenital syndromic monorchism was most probably reported by Chaudhury et al (2010) from India who reported a neonate who had abdominal distension with no passage of meconium caused by ano-rectal malformation which was associated with agenesis of the left kidney and right-side anorchia. Laparotomy showed congenital pouch colon [14].
Unilateral anorchia (monorchism), congenital absence of one testis is an extremely rare condition, and the syndromic form was reported only in two patients. This paper reported the novel association of Goldberg Shprintzen syndrome with monorchism, and this case represented the third case of congenital syndromic monorchism in the world.
The author would like to express his gratitude for the parents who accepted the publication of the photos of their child.
Figure-1A was included in author’s previous publication, but the author has its copyright.
Conflict of interest: None.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
