
Case Report | DOI: https://doi.org/10.31579/2642-973X/087
Assistant Professor in Language Pathology, Department of Speech-Language Pathology, AIISH Mysore.
*Corresponding Author: Si Ahmed Hakim, Assistant Professor in Language Pathology, Department of Speech-Language Pathology, AIISH Mysore.
Citation: Si A. Hakim, Daoudi Smail, (2023), Gliomatosis Mimicking Encephalitis, J. Brain and Neurological Disorders. 6(7):DOI:10.31579/2642-973X/087
Copyright: © 2023, Si Ahmed Hakim. This is an open-access article distributed under the terms of The Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 27 November 2023 | Accepted: 06 December 2023 | Published: 15 December 2023
Keywords: cognitive linguistic deficit; lexical access; task complexity; organisation
Cerebral gliomatosis is a rare glial tumour which is defined by a diffuse and not very destructive infiltration of the encephalon by the glial neoplastic cells in the absence of individualizable tumour mass (Sanson and al., 2005).
The clinical and radiological presentation is often misleading and not very specific, and the diagnosis is rarely mentioned. Histological diagnosis remains difficult. Finally, gliomatosis poses a specific therapeutic problem compared to other glial tumours due to the toxicity of panencephalic radiotherapy and the impossibility of achieving surgical reduction of the tumour (Sanson and al., 2005).
Data from the literature show a median overall survival of 14.5 months, a higher frequency of oligodendroglial forms. The prognosis is linked to age, functional status, histological grade, oligodendroglial differentiation (Sanson and al., 2005).
We report the observation of a gliomatosis occurring in a 14-year-old boy, having presented focal subintral epileptic attacks accompanied by hemiparesis. Flair sequence brain MRI showed a left fronto-temporo-insular hyper signal. The brain biopsy revealed gliomatosis. The evolution was favourable after radiotherapy. Gliomatosis is a diagnosis to be systematically evoked in the presence of a diffuse cerebral affection. Its etiopathogenic mechanism is unknown, and evolution is unpredictable (Millan. BS and al., 2010).
Cerebral gliomatosis is a condition initially described by Nevin in 1938 (Nevin and al., 1938), characterized by diffuse infiltration of glial tumor cells invading a large part of the brain, bilaterally (possibility also of the marrow), with absence of individualizable tumor mass (Nevin and al., 1938).
It is a rare neurosurgical pathology, since less than 300 cases are reported in the literature (Sanson and al., 2005), which can occur at any age, more frequently in adults between 40 and 50 years (MILLAN. BS and al., 2010). The clinical signs are non-specific and the imagery is often misleading, which can simulate a large number of non-neoplastic neurological medical pathologies (inflammatory or infectious encephalitis, angeitis, leukodystrophies) (Sanson and al., 2005). Its radiological and histological diagnosis is difficult because of the absence of identifiable tumor mass and its diffuse character. (MILLAN. BS and al., 2010). Gliomatosis also poses a problem of specific therapeutic management by comparing them to other glial tumors (its diffuse nature, surgery is excluded, radiotherapy is poorly tolerated since it often involves the entire brain, chemotherapy is possible unlike other gliomas) (Sanson and al., 2005).
We report a case of gliomatosis in a 14-year-old boy, diagnosed in pre-mortem using MRI and brain biopsy data, the clinical and radiological picture of which was misleading, simulating an acute limbic encephalitis picture.
A 14-year-old boy immediately developed serial, drug-resistant focal seizures with temporal spread, and then developed right hemiparesis. The examination showed right hemiparesis, with no disturbance of consciousness, no signs of intracranial hypertension, or meningeal syndrome.
Magnetic resonance imaging (MRI) revealed a hyper signal on the Flair and T2 sequences, without contrast enhancement, unilateral, on the left, touching the fronto-temporo-insular white matter (Fig. 1). The GES highlighted left hemispherical brain pain, without paroxysms (Figure 2). The cytochemical study of the CSF revealed a slight hyperproteinorachia at 0.5 g / l, as well as a lymphocytosis at 30 elements / mm3, and the immunological study did not show a chronic inflammatory process. All serologies were negative (Herpes, HTLV1, syphillis, HIV, Lyme ...), and the autoimunity balance was also negative.
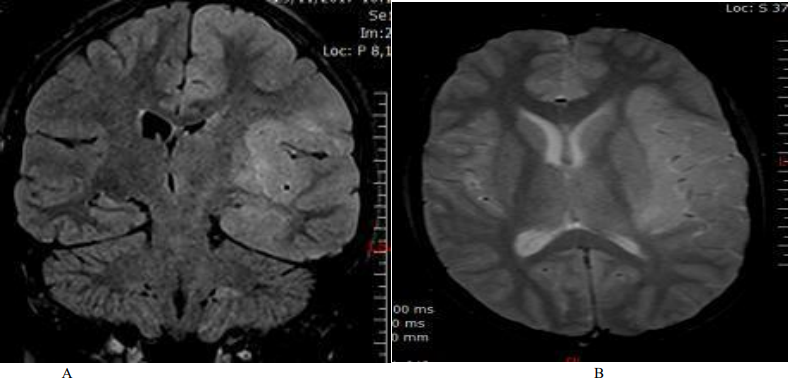
Figure 1. Cerebral MRI: A: coronal section, Flair sequence / B: transverse section, T2 sequence: left fronto-temporo-insular hyper signal.
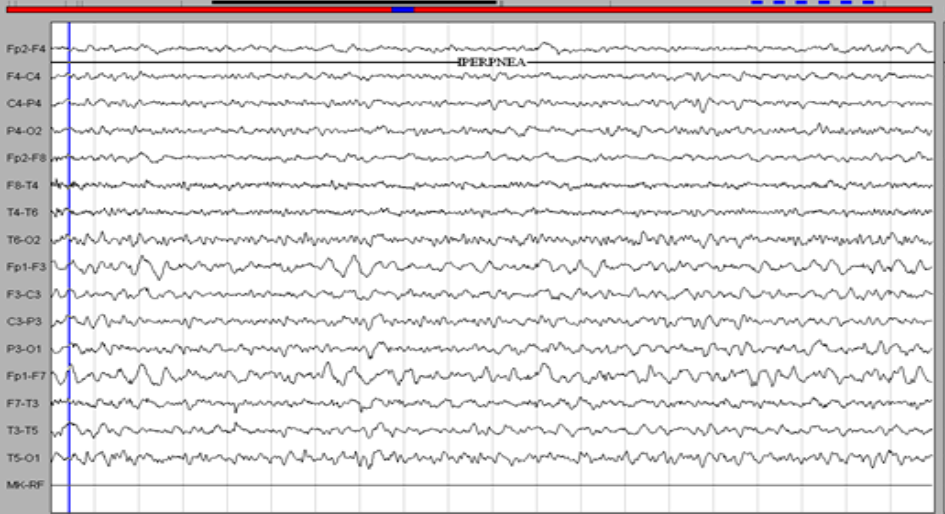
Figure 2. EEG. Left hemispherical pain (theta delta waves more marked on the left anterior regions).
The patient received Methylprednisolone IV (1g / day for 5 days) with an oral relay (1mg / kg / day), combined with anti-epileptics (Levetiracetam 3g / day, Lamotrigine 200mg / day). The evolution was marked by a persistence of the epilepsy attacks, an appearance of headache and oculomotor disorders (diplopia, left exophthalmia), and see obnubilation. Control MRI showed an aggravation of the lesions with mass effect and bilateralization of the hyper signal (Figure. 3). The brain biopsy was done urgently and returned in favor of a gliomatosis (oligodendrogliale). The child was treated with radiotherapy, with disappearance of disturbances of consciousness and seizures, and persistence of hemiparesis.
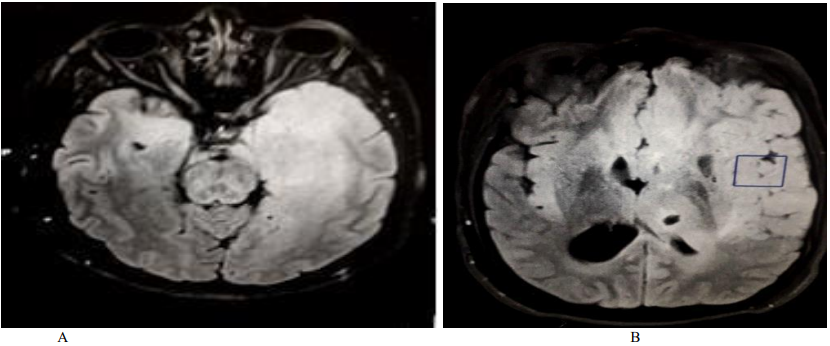
Figure 3. Control brain MRI (FLAIR sequences, cross section): extensive hyper signal affecting the fronto-temporo insular white matter, bilaterally, with mass effect.
We report a case of cerebral gliomatosis, the diagnosis of which was based on imaging and brain biopsy data and whose evolution was favorable with disappearance of epilepsy attacks, improvement in disorders of consciousness and hemiparesis.
Primary gliomatosis often poses diagnostic difficulties because its clinical presentation is not very specific. The most frequent modes of revelation were epilepsy, the occurrence of cognitive disorders, intracranial hypertension (headache), the presence of focal deficits (ARTIGAS. J, and al., 1985, Sanson and al., 2005). It was in 1986 that the first premortem diagnosis of gliomatosis was made on the combined brain biopsy and MRI data (Troost D, and al., 1987). The incidence is higher in humans (in fact common to all gliomas) (Sanson and al., 2005) and, more specifically in gliomatosis, a younger age of onset in humans (Jennings and al., 1995).
In MRI, the Flair and T2 sequences are essential, they show an extended bilateral hyper signal of white matter and gray nuclei, which can extend to the brainstem and the spinal cord (Peretti-Viton P, and al., 2002 ), and gadolinium intake is generally absent (Sanson and al., 2005). It is defined as "an infiltrating process, encompassing at least three lobes, without contrast enhancement or less than 1 cm" (Sanson and al., 2004). The brain scan, including with injection, may be normal, but a careful examination may show suggestive abnormalities (discreet ventricular asymmetry, discreet poorly limited hypodensity, appearance of diffuse cerebral edema with small ventricles, and diffuse erasure of the furrows, or, in an elderly patient, the absence of atrophy of the brain scanner) (Sanson and al., 2005). The diagnoses most often mentioned were: multiple sclerosis, leukoencephalopathy of unknown cause, encephalitis, progressive multifocal leukoencephalitis, vasculitis, Behçet's disease (Sanson and al., 2005). Although not specific, certain aspects are suggestive of gliomatosis: the presence of a discrete mass effect, the thickening of the corpus callosum, the asymmetrical character of the hypersignal, the heterogeneous and "flaky" character of the hypersignal, better visible in T2 as in FLAIR, the associated impairment of the gray matter, in particular of the thalamus and the loss of the boundaries between the white matter and the gray matter (Sanson and al., 2005).
The clinical and radiological differential diagnosis includes all diffuse encephalopathies of various causes (infectious, vascular metabolic and tumor) (Peretti-Viton P, and al., 2002). This is the case of our patient who presented a misleading picture with clinical and para-clinical signs (MRI, EEG, CSF) of acute limbic encephalitis.
The diagnosis by histological study by biopsy is still essential, but often proves difficult, and sometimes non-contributory due to the low cell density, and the preservation of the normal architecture (Sanson and al., 2005). The histological grade is sometimes difficult to determine (Sanson and al., 2004). (astrocytic, oligodendroglial, polymorphic aspect…). According to the literature, oligodendroglial gliomatosis is considered rare (Balko and al., 1992), unlike some authors where the majority of gliomatosis is of oligodendroglial type (Sanson and al., 2005), as was observed in our patient. The extremely diffuse aspect of tumor infiltration could explain the fact that the brain biopsy is sometimes not very contributory. Glioma cells do not have the capacity to cross the vascular basement membrane (limiting metastases by hematogenous or lymphatic route) (Tonn and al., 2003), but they have the capacity to migrate very far into the neuron (Tonn and al., 2003; Bellail and al., 2004).
Spontaneous evolution is extremely variable ranging from less than 1 month to 16 years (Artigas, 1985, Cervos-Navarro, 1987, Louis. DN, and al., 2007). Survival appears to be linked primarily to clinical factors: gender, age and Karnofsky's functional index. The prognosis also appears to be linked to histological characteristics (Sanson and al., 2005). Our case had a favorable evolution with follow-up for 08 months, marked by a regression of clinical signs (epilepsy attacks, disturbances of consciousness, oculomotor disorders). The criteria which seem in favor of a good evolution are criteria of clinical response (disappearance of convulsions, regression of cognitive disorders and headache) or radiological (reduction of the range of the hyper signal, regression of the mass effect) (Sanson and al., 2004, 2005).
Exeresis is not possible because of the extent of gliomatosis (Sanson and al., 2005), and radiotherapy has a major neurotoxicity, since it often involves the entire brain (Crossen and al., 1994). This is why some authors have proposed treating these patients with chemotherapy alone as first-line treatment, which has the advantage of better tolerance (Sanson and al., 2004).
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
