
Review Article | DOI: https://doi.org/10.31579/2640-1053/214
1Riggs Pharmaceuticals, Department of Pharmacy, University of Karachi.
2Pharmaceutical Inc OPJS University Rajasthan.
3Assistant Professor, Dow University of Health Sciences, Karachi Pakistan.
4Associate Professor, Department of Pathology, Dow University of Health Sciences, Karachi, Pakistan.
*Corresponding Author: Rehan Haider, Riggs Pharmaceuticals, Department of Pharmacy, University of Karachi.
Citation: Rehan Haider, (2024), Genetically Engineered Mouse Model in Preclinical Anti-cancer Drug improvement, J Cancer Research and Cellular Therapeutics, 8(7); DOI:10.31579/2640-1053/214
Copyright: © 2024, Rehan Haider. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 08 October 2024 | Accepted: 22 October 2024 | Published: 04 November 2024
Keywords: genetically engineered mouse model (gemms), cancers drug development, preclinical studies, tumor microenvironment, pharmacokinetics, pharmacodynamics, drug resistance, personalized medicinal drug
Genetically engineered mouse fashions (GEMMs) have grown to be essential equipment in preclinical anti-most cancers drug development. Those fashions are designed to duplicate precise genetic alterations discovered in human cancers, providing a more accurate illustration of tumor biology and therapeutic responses. GEMMs allow for the look at tumor initiation, development, and metastasis inside a physiologically relevant microenvironment, offering insights into mechanisms of drug resistance and cancer evolution. Importantly, GEMMs assist in evaluating novel therapeutics' efficacy and protection, mimicking human pharmacokinetics and pharmacodynamics more intently than traditional xenograft fashions. Their use helps the identity of predictive biomarkers, permitting a more personalized method for cancer remedy. Moreover, GEMMs are treasured for testing combination treatment plans, assessing capability synergistic consequences, and understanding the tumor's immune panorama in immunotherapy research. No matter their blessings, GEMMs face barriers, consisting of high costs and time-extensive improvement. Additionally, no longer all cancer mutations are difficult to replicate in mice. Nonetheless, persistent advances in genetic engineering techniques, including CRISPR/Cas9, are increasing the application of GEMMs in oncology studies. Via improving the translational relevance of preclinical research, GEMMs play a pivotal function in accelerating the invention and development of more powerful anti-most cancer treatment plans, in the end improving patient results.
In step with the maximum recent American Most Cancers Society records, an envisioned 569,490 people died from cancer in 2010 (American Cancer Society [ACS], 2010[1]. The wide variety of most cancers-associated deaths currently handed the ones from coronary heart disorder in Americans <85 years vintage (Kung et al., 2008)[2]. Developing new and greater efficacious anti-most cancer compounds is a paramount health care priority. At the Developmental Therapeutics program of the U.S. Countrywide Cancer Institute, ability therapeutic marketers are commonly tested for interest in an in vitro 60-tumor cell line screen and sooner or later using in vivo xenograft research in rodents (Fig 1). Once decided on for additional trying out the use of mounted standards (drugability, the novelty of structure and/or mechanism of movement, efficiency, mobile panel selectivity, and many others.), extra studies (pharmacokinetics, pharmacodynamics, variety finding toxicity, method, mechanism of motion analysis, IND-directed toxicology, etc.) are initiated observed by way of the development of a specific few candidate dealers into human clinical trials. Efficacious capsules with tolerable toxicity are ushered into early-phase scientific trials. However, most of the promising compounds identified in the multi-layered preclinical screenings aren't as successful in human patients. Consequently, preclinical models that better predict drug efficacy and toxicity in humans are wished. Differences in drug absorption, distribution, metabolism, and elimination (ADME), immune responses, and over-interpretation of the preclinical efficacy records may additionally contribute to scientific disasters. But, a widespread hassle is producing mouse models that histologically, genetically, and behaviorally recapitulate human disease Over the last two decades, researchers have identified a number of the underlying genetic abnormalities that cause positive cancers and have genetically engineered them into mouse fashions to explore the oncogenic nature of these genes and related pathways. The resulting mouse fashions generate tumors that could highly mimic those seen in people. It's far expected that healing compounds that show efficacy and coffee toxicity in these more human-like genetically engineered mouse (GEM) models can be more of a hit in scientific trials and cause expanded cancer survivorship. Hundreds of GEM models currently exist that can be related to some form of human tumor development (see the eMICE website). Its miles properly past the scope of this manuscript to adequately describe each of them. We rather in a short overview of some of the floor-breaking GEM models advanced within the Eighties and 90s, describe more recent and complex models, spotlight a number of the advantages and barriers for every, and discover the potential roles of molecular-centered treatment plans and GEM models in preclinical drug trying out. For added info on each version, we refer to the reader to the original articles or previous indepth opinions. Whilst mice that increase genetically engineered tumors preserve a whole lot of promise, the tremendously gradual increase in the price of those tumors and their low penetrance (no matter genetics, a few mice will no longer broaden tumors all through the time constraints of the experiment) makes them sick-suited to update xenograft fashions as the primary in vivo screening device. Alternatively, we envision GEM fashions as a further screening the mechanism that narrows and optimizes the sphere of therapeutic compounds earlier than luxurious and timeconsuming human clinical trials are initiated.

Figure 1: Wherein gemstones may be maximum useful inside the Preclinical Drug development.
Simplistic assessment of preclinical drug development manner within the NCI/DTP. Novel dealers are brought via the 60 cell Line screen (reviewed in Priests et al., 1991), which incorporates 60 human-cultured tumor cellular lines in a 96-well plate array. If anti-proliferative activity is observed in several mobile lines, the agent is examined in a hollow Fiber assay, which measures the anti-proliferative hobby in cultured cells grown in hole polyvinylidene fluoride (PVDF) fibers implanted intraperitoneal and subcutaneously in mice (reviewed in Decker et al., 2004). If lively in hollow Fiber assays, dealers are administered to immunodeficient animals that possess human tumors generated from serially passaged human tumor mobile lines (xenografts). Tolerable toxicity and interest are measurable in xenograft models leading to simultaneous investigations into the agent’s/PD, mechanism of movement, and toxicology. Based totally on this information, wherein applicable an IND utility is generated for FDA approval to initiate scientific trials. The sellers may also require further preclinical studies or can be permitted for a small-scale segment 0 trial (reviewed in Murgo et al., 2008), or preliminary segment I human scientific trials. Assay Time is a satisfactory case state of affairs for a single agent and no longer consists of time for optimization and validation repetitions. PD = pharmacodynamics, PK = pharmacokinetics, GEM = genetically engineered mouse, IND = Investigational New Drug.
After a potential healing agent suggests a few interests in cultured tumor cell growth Assays, its activity and toxicity are explored through in vivo xenograft models. Fragments of human tumors or cultured human tumor cells are implanted (often subcutaneously) into immunodeficient rodents. As soon as the tumors develop to a predetermined length, the capability antimost cancer agents are administered and the tumor response, generally assessed by way of monitoring the tumor size, is measured through the years. Drug doses and management schedules are adjusted to optimize efficacy and decrease toxicity over numerous experimental cycles. This system generates valuable records and fast returns a prediction on an agent’s pastime, regularly for numerous months. Lamentably, promising anti-cancer compounds located through this route often fail in human scientific trials, typically due to low efficacy [3,4]. The low predictive cost of these xenograft models exemplifies the want for better in vivo mouse models for preclinical drug checking out.
The transgenic mouse era has advanced from the early 1980s [5] such that researchers can now conditionally and reversibly alter single gene expression. Thousands of publications have supported the speculation that oncogene expression or tumor suppressor gene ablation in everyday mammalian cells is sufficient to pressure tumor development. The first file of a heritable tumor-susceptible transgenic mouse came here in June 1984 (Brinster et al., 1984[6]. Those mice used the SV40 enhancer location to assist force expression of a construct including the mouse metallothionein-1 gene promoter fused to the thymidine kinase gene from the herpes simplex virus (HSV). They blanketed the complete SV40 upstream location, along with enhancer, promoter, and two T-antigen genes transcribed inside the contrary direction. It became the notion that T-antigen genes would be inactive in mice. , they observed that the transgenic mice constantly advanced brain tumors, in addition to sporadic tumors in other tissues. Comply with-up experiments confirmed that the use of only the SV40 enhancer/promoter place and the massive T-antigen gene becomes sufficient to pressure tumorigenesis [7]. Next reports have shown that expressing the large T-antigen in a particular cellular type can sell tumor development. For example, Ornitz et al., (1985) established expression of the massive T-antigen in acinar cells generated exocrine pancreatic tumors, whereas Hanahan (1985) used the insulin promoter using the large T-antigen particularly in pancreatic beta-cells to produce endocrine pancreatic tumors. Following the sudden oncogenic capability of SV40, researchers attempted to rationally design a tumor-generating transgenic mouse (“oncomouse”). So that you can create a mouse version of a chromosomal translocation visible in some human B-cellular lymphomas, [8] generated a transgene consisting of an immunoglobulin enhancer (E) driving expression of the Myc gene. Those mice heritably expand pre-B cellular and mature B-cellular lymphomas. Further research through Strasser et al., (1990) showed that Myc required extended expression of the anti-apoptotic thing Bcl-2 to force tumorigenesis. Suppression of apoptosis is now understood to be a hallmark of many cancer cells.
3.1 MMTV precipitated breast cancer
the use of reciprocal matings between excessive tumor and occasional tumor mouse traces, Bittner (1936) suggested tumor prevalence in F1 girls became dependent on the stress of the mom. Virologists validated a plague (dubbed Murine Mammary Tumor Virus, MMTV) was chargeable for inducing tumors in mammary tissue and becoming handed from mom to offspring through her milk. Next studies showed some mouse strains additionally had MMTV virus in their eggs and sperm (reviewed in Heston & Parks, 1977).[9] The application of MMTV was expanded when it became proven that a short regulatory region (known as long terminal repeat, LTR) became enough to confer hormone-responsive and cellular-specific expression in vitro [10]. Stewart (1984)[11] used the MMTV LTR to force Myc expression in his transgenic mice that advanced breast adenocarcinomas in mammary epithelial tissue. When you consider that then the MMTV LTR has been fused with an expansion of purported oncogenes to expand tumors in murine mammary tissue which are comparable in morphology and gene expression profile to certain varieties of human breast cancers [12]. For example, the MMTV-driven Polyoma middle-T antigen (PyMT) the model develops tumors much like a human breast most cancers with luminal-type morphology approximately 2-3 months after beginning (Man et al. 1992). A version expressing MMTV-pushed Wnt1 (wingless-kind MMTV integration website own family, member 1) generates mouse mammary tumors with characteristics similar to those of human basal-kind breast cancers [13] several members of the Wnt gene circle of relatives (encoding secretory glycoproteins that generally stimulate cellular proliferation and differentiation) are expressed inside the mouse mammary tissue all through diverse tiers of development. Wnt-1 isn't usually expressed in the mammary tissue; however, while driven ectopically by using MMTV, it develops oncogenic houses. Like most of the opposite “first generation” transgenic oncomice, many of these models have a low tumor penetrance and broadly variable latency length, making them tough to apply directly in large-scale preclinical drug screenings. These obstacles are partially conquered by resecting and transplanting transgenically brought tumors into many syngeneic recipient animals, generating a large cohort of tumor-bearing animals for drugscreening functions [14,15]
3.2 Activated Kras
The KRAS2 gene encodes a G-protein that is a mammalian cellular homolog of a transforming gene isolated from the Kirsten RAt Sarcoma virus. This membrane-associated intracellular signal transducer performs a vital position in ordinary tissue signaling, proliferation, and differentiation [16]. Numerous oncogenic point mutations intervene with the intrinsic GTPase interest of Kras, causing accumulation in a constitutively lively GTP-sure nation.[17] Expressing an activated Kras mutant transgene in acinar cells induces neoplasia in the fetal pancreas with massive tumors developing only days after pancreatic differentiation. Certainly, activating factor mutations within the Kras gene has finally been proven to arise in 75 to 95% of spontaneous human pancreatic cancers [18] in addition to > 90% of spontaneous and chemically caused mouse lung tumors [19]. Activated Kras expression inside the mouse lung generates more than one tumor at an early age, so much so that the mice succumb fast due to respiration failure [20] The numerous penetrance and multi-focal primary tumor formation further to the fast lifestyle span limits the usage of this version in addition to studies of tumor development.
3.3 Knockout oncomice
The examples described thus far depend on the over-expression of a nucleic acid sequence with purported oncogenic houses or mutations to pressure tumorigenesis in mice. at some point in the late 1980’s using homologous recombination in mouse embryonic stem cells enabled researchers to inactivate (“knockout”) unmarried genes. This era created a new wave of transgenic oncomice starting with the heterozygous null retinoblastoma (Rb) mouse [21]. Rb inhibits the cell cycle by way of repressing the expression of genes required for S phase progression (reviewed in Hanahan & Weinberg 2000). Mice missing one Rb allele increase pituitary adenomas, while RB null offspring fail to increase past embryonic day 14 or 15, in all likelihood due to immoderate neuronal cellular loss of life. The significance of tumor suppressor expression has become obvious from this and different knockout mouse models. Diverse mobile stresses prompt p53 to modulate the expression of its target genes, lots of which adjust mobile cycle arrest, apoptosis, DNA repair, or cellular metabolism. Reduced or null expression of p53 has been found in several human cancers (reviewed in Harris & Hollstein 1993). [22] Mice missing one or each p53 alleles are born every day however are predisposed to growing spontaneous lymphomas and sarcomas later in life [23]. missing a chief tumor suppressor pathway, these p53 null mice have become a useful heritage with which to clarify the oncogenic capability of other genes. The Rb/p53 double knockout mice expand relatively competitive tumors inside the cerebellum seen as early as 7 weeks of age.[24] Mammary and skin tumors increase frequently in woman mice sporting conditional null Brca2 and p53 alleles, [25] suggesting synergistic inactivation of Brca2 and p53 can mediate mammary tumorigenesis. When you consider that these early transgenic and knockout mouse models have genetic changes that are expressed within the germline and maximum, if no longer all, somatic cells, the models are more consultant of human cancer predisposition syndromes. This isn't always the case for most human cancers, which broaden spontaneously in a small number of cells inside the grownup. Many genes have distinct capabilities and expression styles in the course of embryonic improvement that are nevertheless poorly understood and extraordinary from those inside the adult. Furthermore, those early transgenic fashions are notorious for their variable penetrance and tumor latency, making it almost possible to accumulate the variety of animals with synchronous tumor improvement wished for large research. While large-scale manufacturing of transgenic mice is now viable with IVF and other excessive manufacturing methods (JAX® pace enlargement provider; Charles River Laboratories' fast growth offerings), no longer obviate the problem of variable penetrance and tumor latency; it truly offers a massive range of animals that can be held concurrently to have a look at for tumor development. Early GEM models generated a couple of primary lesions which a long way exceeded that found in human beings, limiting their predictive value and usefulness for preclinical studies. To better model maximum human cancers, genomic alterations have to arise in a small variety of cells in the grownup mouse tissue similar to the microenvironment wherein human cancer develops
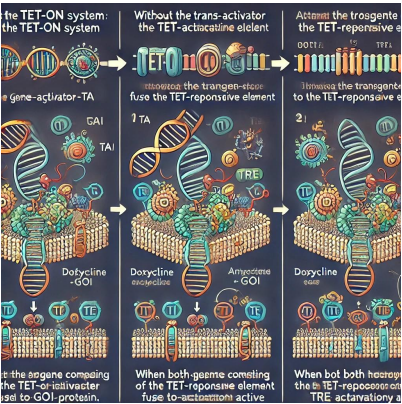
Figure 2: Schematic representation of the Tet-ON system; 1) Without the Tet-Activator (TA), the transgene such as the Gene-of-interest (GOI) fused to the Tet-Responsive element (TRE) will no longer be expressed. 2) Doxycycline can bind to the Tet-Activator protein, altering its protein shape such that TA can now bind to a TRE. 3) If both the Tet Activator and the TRE-GOI transgenes are present inside the identical cellular, upon doxycycline management and TA activation, the GOI can be transcriptionally active.
Taking advantage of the Lox-forestall-Lox conditional transgene expression system, Jackson et al., (2001) created a transgenic mouse version that replaces one wild-type Kras allele with a transcriptionally silent oncogenic Kras-G12D allele. Intranasally introduced adenovirus containing the Cre-recombinase (adeno-Cre) splices out the LSL and allows expression of the activated Kras-G12D transgene inside the lung. Small lesions may be seen 2 weeks after induction; by 12 weeks after induction, adenomas are found, a few with cytogenic characteristics of malignancy. With the aid of 16 weeks of submit induction, huge adenomas and adenocarcinomas are found in many animals. Virus titrations in some of animals demonstrate that the quantity of virus particle equivalents used for induction is at once associated with the quantity of tumors that broaden. Across the equal time, Fisher et al., (2001) created an activated Kras Tet-ON/OFF inducible version. Doxycycline (DOX) administration induces expression of the murine oncogenic Kras G12D allele in alveolar type II pneumocytes. Hyperplasia is determined after just seven days of DOX management; at 8 weeks submit induction, adenomas, and adenocarcinomas are discovered inside the lungs. Removal of DOX from their weight loss program reasons a speedy decrease in activated Kras [removed]inside seven days) and apoptotic regression of the tumors. One month after DOX withdrawal, lesions, and tumors were not found in the lungs, implying expression of the mutant Kras gene product became required to hold the viability of tumor cells. The requirement of persisted oncogene [removed]or regular inhibition of tumor suppressor) to maintain the brought about tumors has given upward thrust to the theory of “oncogene addiction” (reviewed in Weinstein, 2002).
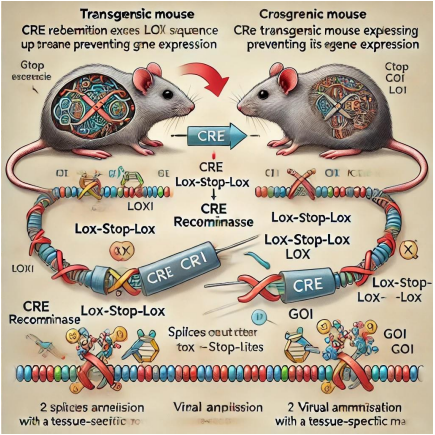
Figure 3: CRE Recombination Mediates Lox-prevent-Lox Gene Activation. One popular approach of inducing gene expression in person mice is to use CRE recombination. 1) The transgenic mouse initially has the Lox-stop-Lox collection inserted upstream of the transcription start website online for the gene of interest (GOI). This prevents GOI expression until acted upon via CRE.
CRE recombinase is added to the correct cell population either with the aid of crossing with a 2d transgenic mouse expressing CRE recombinase the usage of a tissue precise promoter, or using administering a virulent disease expressing CRE recombinase in a tissue-specific manner. 2) CRE recombinase splices out the prevent collection between the 2 loxP sites from the transgenic DNA sequence, allowing GOI expression Sos et al., (2009) currently used computational genomic analysis to become aware of molecular and genetic predictors of therapeutic reaction to clinically relevant compounds in various NSCLC most cancers cell lines which can be highly consultant of primary tumors. Certainly, one of their findings was that cells expressing activated Kras also had more suitable Hsp90 dependency. They used the LSL-KrasG12D model to verify this remark. Mice have been, to begin imaged by MRI for 12-20 weeks put up adeno-Cre administration, then broken up into placebo and 17(Dimethylaminoethylamino)-17-demethoxygeldanamycin (17-DMAG a well-known Hsp90 inhibitor) organizations. Mice were imaged via MRI after 1 week of drug remedy; tumor extent was reduced through 50% in 17-DMAG treated mice even as tumor volume accelerated barely in placebo dealt with animals. Before this look, there were no molecular-centered treatments for activated oncogenic Kras-driven lung tumors. However, by combining the strength of computational genomics and GEM models, researchers were able to rationally perceive a viable healing modality and compare its efficacy in vivo
4.2 Inducible oncogenic mutations within the EGF receptor
The epidermal growth component receptor (EGFr) is a transmembrane mobile surface receptor which is mutated or instead expressed in a sizable proportion of human gliomas and adenocarcinomas (in particular of the head and neck, breast, colon, lung, and pancreas). EGFr is the human homolog of neu, a rat gene first identified in ethyl nitrosourea that brought about tumors (reviewed in Maguire & Greene, 1989), and is a member of the erythroblastic leukemia viral oncogene/human epidermal increase aspect receptor (ERBB/HER) receptor tyrosine kinase own family. Of the most generally found EGFr mutations visible in lung adenocarcinomas encompass an in-body deletion in exon 19 (removing an L-R-E-A [leucine-arginine-glutamic acid-alanine) amino acid motif which is conserved in all regarded vertebrate EGFr sequences) and a factor mutation which results in a leucine to arginine substitution at position 858. these and different much less not unusual mutations inside the kinase domain of EGFr are associated with multiplied therapeutic sensitivity to tyrosine kinase inhibitors along with gefitinib and erlotinib. numerous research tested appreciably prolonged survival and time to development (TTP) in non-small-cell lung, most cancer (NSCLC) patients are treated with gefitinib as their first-line therapeutic (Han et al., 2005; Sequist et al., 2008; Yang et al., 2008). Erlotinib administration in patients who've had previous chemotherapy has a fair more survival gain and is presently recommended by the FDA for 2d line remedy in new NSCLC patients (Shepherd et al., 2005). Inducible transgenic mouse models help to clarify this statement and offer a system to test other therapeutics that function via the EGF receptor. Politi et al., (2006) generated several Tet-ON/OFF mouse systems that overexpress diverse mutant EGFr alleles upon DOX management. Lung adenocarcinomas have been found after months post transgene induction within the exon 19 deletion and L858R transgenic EGFr mouse fashions. Much like the Tet-On/OFF activated Kras gadget, after one month of DOX withdrawal the located tumor load within the EGFr transgene inducible fashions regressed to near pre-induction histology. To check the anti-tumor efficacy of an already clinically accredited healing on this version, the tyrosine kinase inhibitor erlotinib was administered to corporations of transgenic EGFr mice maintained on DOX. After much less than a week of erlotinib remedy, all of the animals that obtained a minimum of 12 mg/kg/d confirmed partial or entire responses by magnetic resonance imaging (MRI). Longer lengths of treatment correlated with extra tumor regression. Ji et al., (2006) generated similar inducible EGFr transgenic mice (exon 19 in-body deletion or L858R) but also brought a luciferase fusion gene to the assembly. This permits the monitoring of lung tumor increase and regression in vivo with the usage of a non-invasive luciferase imaging device. The potential to non-invasively image tumor load again and again in vivo enables the effects of anti-most cancer drug management to be observed long term because the authors validated with cetuximab..
4.3 Gemstones possessing multiple genetic alterations
Patients with lung adenocarcinomas containing one of the not-unusual EGFr oncogenic mutations (deletion in exon 19 of the kinase area [Del.ex19] and leucine 858 to arginine[L858R]) are touchy to the EGFr-tyrosine kinase inhibitor (TKI) treatment options (gefitinib and erlotinib). But, the TKI dealt with tumors always relapse within 12 months through developing resistance to the TKI therapy (reviewed in Morita et al., 2009), most commonly through one of the following mechanisms: EGFr-T790M point mutation (Kobayashi et al., 2005; Pao et al., 2005), MET proto-oncogene amplification (Engelman et al., 2007), or hyperphosphorylation and activation of insulin-like growth issue-I receptor (Guix et al., 2008). The EGFr-T790M mutation (observed in 50% of relapsed EGFr-mutant tumors) is the notion to increase the ATP-binding affinity using an order of magnitude (Yun et al., 2008), thereby reducing the healing ability of TKIs to out-compete ATP for receptor binding. MET amplification is a concept to induce gefitinib resistance generally by using riding ERBB3 (HER3)-dependent activation of the PI3K and Erk pathways, which adjust cellular survival and mitogenesis, respectively (reviewed in Kim & Salgia, 2009). To triumph over the obtained resistance of first technology TKIs, researchers have currently developed TKIs that bind EGFr irreversibly and are presently in or have completed segment II clinical trials (reviewed in Riely, 2008). The pan-ERBB inhibitor HKI-272 covalently binds to a cysteine inside the EGFr kinase area and regressed tumors at nanomolar concentrations in EGFR-L858R transgenic mice (Ji et al., 2006). The capability to inhibit ERBB2 (HER2) has led HKI-272 is to be investigated basically in breast cancer scientific trials (clinicaltrials.gov, 2011). But, different cancers pushed with the aid of ErbB mutations may also be dealt with using HKI-272. Similarly, the irreversibly binding EGFr and HER2 inhibitor BIBW2992 is being tested in over 20 clinical trials of patients with superior NSCLC, breast cancers, prostate cancer, malignant gliomas, and different stable tumors (clinicaltrials.gov, 2011). In addition to ERBB inhibitors, researchers are investigating different druggable targets associated with the EGFr/HER2/ERK/AKT pathways. Shimamura et al., (2008) used an EGFR-L858R-T790M double transgenic GEM model to test the efficacy of an Hsp90 inhibitor, CL-387,785. Hsp90 is a chaperone notion to resource in the proper folding of EGFr; CL-387,785 binds irreversibly to Hsp90 thereby interfering with its chaperone characteristic. Cancer-related lethality in people is regularly because of the invasion of metastatic tumors in tissues a long way from the number one tumor. However, few unmarried transgene GEM models reliably develop metastatic lesions. By crossbreeding the pancreas-precise Cre-mediated activated Kras GEM (which alone generates only pre-malignant neoplasia) and the Ink4a/ARF conditional knockout fashions (no longer result in neoplasia), Aguirre et al., (2003) generated mice that swiftly increase invasive, metastatic ductal pancreatic tumors that are lethal using 11 weeks. This tumor improvement is just like the notably lethal human form of pancreatic ductal adenocarcinoma. Even though the rapid murine mortality prevents long-term research, it may be a beneficial model of deadly human malignant cancers in a brief period of cancer prevention studies. In addition, Ji et al., (2007) crossed a lung-particular activated-Kras GEM with the following tumor suppressor knockout gemstones: Ink4a, p53, or the serine/threonine kinase 11 (Lkb1) knockout. In a number of the twin-knockout mice analyzed, the activated Kras x Lkb1 knockout had the most profound phenotype. Those mice display a significantly improved lung tumor burden, rapid tumor onset, and <50> Winslow et al., (2011) improved on this idea by administering a lentivirus expressing Cre recombinase to a double-floxed GEM model. Upon CRE recombination, activated Kras might be expressed and p53 (flox/flox) would be silenced in certain cells of the grownup lung. Because the lentivirus integrates randomly into the host cellular’s genome, the authors had been in a position to use linkage-mediated PCR (LM-PCR) to generate a unique “fingerprint” for every number one tumor and reveal that “fingerprint” in any next metastases. Even though the virus changed into added at a set time factor and plenty of number one foci appeared to effectively undergo the oncogenic double recombination occasion, only a few number-one foci had been accountable for producing macroscopic metastases outdoor the lung, suggesting different genetic or micro environmental elements manipulate a tumor’s metastatic capacity. This is a great instance of using an inducible, double GEM to model genetic changes visible in lots of human lung cancers and discover the genetics of malignant and non-malignant tumors. Tp53 is not the most effective tumor suppressor being actively investigated in the usage of GEM fashions. The tumor suppressor phosphatase and tensin homolog (PTEN) is a lipid phosphatase that negatively regulates the PI3/AKT pathway, which is often upregulated in human malignancies. Sixty percent of primary human prostate cancers show a decrease/lack of PTEN expression; this gene also is deleteriously mutated in 70% of gliomas and in some varieties of breast carcinomas and melanomas (grey et al., 1998). To research the role of PTEN in prostate cancer, Trotman et al., (2003) created numerous PTEN GEM models, every with an exceptional stage of PTEN expression. Simplest the homozygous PTEN knockout displays an enormous phenotype (invasive prostate cancers develop after six months in all mice). However, not one of the models generates metastases and the mice no longer have reduced survival compared to wild-type controls. A prostate-specific PTEN conditional knockout was created which also generated invasive prostate cancer; but in this case, metastatic prostate tumors evolved and were discovered in the lymph and lung (Wang et al., 2003). Concomitant inactivation of one or each Cdkn1b (encodes the tumor suppressor p27) alleles in a heterozygous PTEN knockout mouse accelerates spontaneous neoplasia formation which develops into prostate carcinoma at complete penetrance within 3 months from birth (Di Cristofano et al., 2001). While a conditional PTEN knockout mouse is crossed with a p53 knockout mouse the resulting offspring is a conditional double knockout GEM that develops aggressive, deadly prostate most cancers with whole penetrance in seven months (Chen et al., 2005). The cancers discovered in this new era of GEM models recapitulate the progression and histopathologic capabilities of some forms of prostate cancer determined in people. This research spotlights the function of PTEN in tumorigenesis and the want for additional cooperative tumor suppressor inactivation to generate malignancies and deadly cancers. The capacity for drug discovery and validation of the usage of conditional GEM fashions is confirmed with the Brca1/p53 mouse version. Most people with human BRCA1-associated tumors harbor mutations in both p53 and BRCA1. Poole et al., (2006) created any such CreLoxP conditional GEM model via inactivating both p53 and Brca1, especially inside the mouse mammary gland. The authors found that progesterone receptors are overexpressed in mutant mammary epithelial cells and provided a possible road of treatment. Administration of a progesterone agonist (mifepristone, RU 486) prevented mammary tumorigenesis in Brca1/p53-poor mice. as the potential to genotype a patient’s tumor to determine their status for EGFr, MET, p53, IGF, and different essential neoplasia-associated objectives turn into realistic, it is going to be feasible to higher pick the finest mixture therapy to impact the applicable oncogenic pathways.
The ability to predict the energy of transgenic mice can be illustrated in the instance of thiazolidinedione’s (TZDs). As an agonist of peroxisome proliferator-activated receptor gamma (PPAR), the TZD troglitazone was an FDA-accredited therapy for type-2 diabetes. TZD anti-tumor activity in cultured and xenografted human colon cancer cells brought about exhilaration inside the scientific community that initiated its use in phase II clinical trials in sufferers of colon cancer (Sarraf et al., 1998). However, in a look posted a few pages after the xenograft look, troglitazone showed no anti-tumor pastime in Min+/- mice; polyp formation extended with troglitazone administration in this model (Saez et al., 1998). This heterozygous knockout mouse model lacks one purposeful replica of the APC tumor suppressor gene, for this reason, predisposing them to colon cancer. Inside the section II medical trial patients who dealt with troglitazone honestly confirmed disorder development within months of remedy initiation, correlating with the outcomes anticipated from the transgenic mouse model (Kulke et al., 2002).
5.1 Cautionary Tale
Even though many regard mice as “little fuzzy people” in the evaluation of preclinical statistics, there are tremendous and often unknown differences in drug metabolism, gene expression, and ailment development among the murine version and people. One example of transgenic mouse models failing to as it should be predicted human scientific final results is the Farnesyltransferase (FTase) tale. FTase catalyzes the publish-translational farnesylation of proteins containing a C-terminal CAAX motif, wherein C is the cysteine residue to be farnesylated, A represents an aliphatic amino acid, and X is any amino acid. Whilst X is leucine, the protein is a preferred substrate for the same enzyme named geranylgeranyltransferase I (GGTase).
Mendola & Backer, (1990) showed farnesylation of Ras proteins is important for their Transformation into amazing oncogenes. This discovery ignited the speculation that interfering with crenulation the use of inhibitors of FTase (FTIs) or GGTase (GGTIs) should cause tumor growth inhibition and a feasible anti-cancer therapy. Even though some facts recommend an FTI and a GGTI wants to be applied simultaneously to obtain whole inhibition of Kras prenylation (Rowell et al., 1997), FTIs on my own have been shown to inhibit the growth of human tumor xenografts in nude mice (sun et al., 1998). FTI monotherapy also demonstrates beneficial results in transgenic mouse fashions: tumor regression in H-Ras mice (Kohl et al., 1995), tumor stasis in N-Ras mice (Mangues et al., 1998), and tumor growth inhibition in Ki RasB mice (Omer et al., 2000). Those consequences and others spurred pleasure for FTI monotherapy in scientific trials. Excluding some trials in breast cancer and leukemia patients, FTIs used as single sellers have no longer proven true efficacy towards strong tumors (reviewed in Zhu et al., 2003). Therefore, not one of the numerous mouse fashions evaluated has been capable of predicting drug efficacy in people. However, many scientific trials are currently underway combining FTIs with different tablets such as “classical” anticancer remedies (chemotherapy or radiotherapy) and molecular-centered therapeutics
5.2 Targeted Molecular Therapeutics
For decades, cancer therapy has been one or an aggregate of the subsequent three treatments surgical operation, radiotherapy, and cytotoxic chemotherapy. However, all have their obstacles and considerable facet results. Ideally, anti-most cancer treatments could “goal” cancer cells and decrease detrimental effects on non-cancerous cells. Tumors frequently incorporate gene expression profiles and mutations that make them more or less sensitive to some focused therapies than others. Identifying the tumor’s gene expression profile and correlating it to the maximum successful remedies for that particular profile will hold healing damaging side consequences to a minimum and yield a great diagnosis for the patient.
The 2 major forms of centered molecular treatment options are monoclonal antibodies, which target the receptor’s extracellular domain, and small molecule inhibitors, which goal the intracellular signaling and kinase domains. Table 1 outlines a number of the FDA-approved focused therapeutics, their molecular targets, and the varieties of cancer wherein they have been demonstrated to have a scientific advantage. Monoclonal antibodies (MAbs) are normally administered no more than as soon as weekly through intravenous injection, whereas small molecules (nibs) can normally be taken orally every day. Currently, targeted healing procedures can be administered as unmarried marketers, however, many display greater blessings in mixture with other sellers or similarly to traditional treatment options in those patient populations with tumors of susceptible genetic profiles. Tumors can be generated in genetically engineered mice that have these inclined mutations and expression profiles, supplying researchers with precious preclinical drug screening opportunities. Herceptin (HER2) is a transmembrane receptor that is over-expressed in 20-25% of breast cancers and is related to aggressive tumor conduct and poor analysis (reviewed in Nanda, 2007). Two early research have proven the anti-HER2 monoclonal antibody trastuzumab plus chemotherapy considerably improves average survival in HER2+ patients with metastatic breast cancer (Slamon et al., 2005). Antibody binding is thought to inhibit HER2 signaling, which disrupts DNA restore mechanisms and induces cytotoxicity. Resistance to trastuzumab typically occurs and may render this treatment useless in subsequent relapses. A current strength has been to conjugate the antibody with a toxin/drug, thereby developing a “guided missile” that goals a specific epitope over-expressed on most cancer cells and delivers the poisonous agent to the one's particular cells. Researchers at Genetech harvested mammary tumors from MMTV-HER2 transgenic mice, implanted them orthotopically into a big cohort of nude mice, staged the mice when suggested tumor volumes have been ~a hundred-two hundred mm3, and administered their trastuzumab-DM1 (T-DM1) conjugate at diverse time factors and doses (Jumbe et al., 2010). DM1, also known as maytansine and derived from participants of several tropical plant households, is a robust cytotoxin that irreversibly inhibits tubulin polymerization and arrests cells in the M or G2 phase (Remillard et al., 1975; Rao et al., 1979). because the toxin is especially focused on tumor cells, the
Authors saw no detrimental activities within the mice, with ~50% of the tumors displaying whole regression at doses of 15 mg/kg given once every three weeks. PD/PK measures had been received from the mice and this immunoconjugate remedy is now being evaluated in several scientific trials in sufferers with metastatic breast cancer. several therapeutics have been developed that focus on the EGFr receptor to deal with non-small cells lung most cancers and colorectal cancer, which include cetuximab, panitumumab, erlotinib, and gefitinib. Tumors expressing EGFr with a deletion in exon 19 or sensitizing factor mutation (L858R) in the ATP-binding pocket reply significantly higher to gefitinib than patients with wild-type EGFr (Lynch et al., 2008). Continually, even the tumors with increased drug sensitivity relapse when resistance to gefitinib develops. Extra therapeutics need to be generated to triumph over the inevitable drug resistance.
Table 1: Overview of FDA-Approved Targeted Therapy Compounds
| Drug Name | Molecular Target | Approved Use |
|---|---|---|
| Cetuximab (Erbitux®) | EGFR | Colorectal Cancer |
| Erlotinib (Tarceva®) | EGFR | Non-Small Cell Lung Cancer, Metastatic Pancreatic Cancer |
| Gefitinib (Iressa®) | EGFR | Non-Small Cell Lung Cancer |
| Panitumumab (Vectibix®) | EGFR | Colorectal Cancer |
| Trastuzumab (Herceptin®) | HER2 | Early and Metastatic Breast Cancer |
| Lapatinib (Tykerb®) | HER2 and EGFR | Breast Cancer |
| Bevacizumab (Avastin®) | VEGF | Metastatic Colorectal Cancer, Metastatic Melanoma, Non-Small Cell Lung Cancer |
| Sunitinib (Sutent®) | VEGFR and PDGFR | Metastatic Renal Cell Carcinoma |
| Sorafenib (Nexavar®) | Multi-Targeted Kinase Inhibitor | Metastatic Renal Cell Carcinoma |
| Toceranib (Palladia®) | Multi-Targeted Kinase Inhibitor | Canine Specific Mastocytoma |
| Pazopanib (Votrient®) | VEGFR, PDGFR, cKIT | Advanced Renal Cell Carcinoma |
| Imatinib (Gleevec®) | cKIT, ABL, PDGFR | Chronic Myeloid Leukemia, GIST |
| Dasatinib (Sprycel®) | BCR/ABL, Src Family | Chronic Myeloid Leukemia resistant to prior therapy |
| Alemtuzumab (Campath®) | CD52 on Mature Lymphocytes | Chronic Lymphocytic Leukemia, Cutaneous T-cell Lymphoma |
| Ofatumumab (Arzerra®) | CD20 on B-cells | Chronic Lymphocytic Leukemia |
| Rituximab (MabThera®) | CD20 on B-cells | B-cell Non-Hodgkin's Lymphoma |
| Nilotinib (Tasigna®) | BCR/ABL kinase inhibitor | Philadelphia chromosome positive chronic myeloid leukemia |
| Vandetanib (Zactima®) | VEGFR, EGFR, RET | Metastatic Medullary Thyroid Cancer |
Since vascularization is needed for the increase and health of a tumor, the angiogenesis-associated component VEGF has become a desired goal for therapy. Bevacizumab is an anti-VEGF antibody that has been shown to bind and inhibit VEGF, slowing angiogenesis and helping in stable tumor starvation and elimination (Brekken et al., 2000). Whilst used as unmarried retailers in unsorted cancer sufferers, anti-angiogenic therapeutics do not show a sizeable pastime. However, while used in combination with different treatment options (in particular cytotoxic chemotherapy), and subgroups of sufferers display promise (reviewed in Cabebe, 2007). Bevacizumab, similar to paclitaxel and a platinum cytotoxic agent, is now part of a first-line remedy for newly identified non-small cellular lung cancer. Loads of section II and section III trials using bevacizumab in a mixture with other healing procedures in lots of other cancers are currently ongoing (clinicaltrials.gov, 2011).
5.3 GEM oncomice in most cancer prevention
GEM fashions are starting to recognize their capacity as preclinical models of cancer prevention (briefly reviewed in abate-Shen et al., 2008). In a recent example, Ohashi et al., (2009) used the EGFR-L858R-FLAG transgenic mouse to check the cancer-preventative the ability of gefitinib administration. 20-5 3-week antique transgenic mice have been administered gefitinib, with 5, 5, and 15 mice euthanized at weeks eight, thirteen, and 18, respectively. At termination, the lung tumors have been counted and measured macroscopically and histologically. The ones receiving gefitinib failed to increase adenocarcinomas, while the businesses given automobile manipulation developed hyperplastic regions at 5 weeks and big adenocarcinomas with the aid of 15 weeks. One week after cessation of the 15-week gefitinib administration duration, the transgenic mice showed signs and symptoms of hyperplastic mobile growth in the lung (via PCNA staining), suggesting gefitinib was the lone component preventing EGFrL858R tumor improvement in those mice. long-run research is had to see if the EGFrL858R transgenic mice finally increase gefitinib resistance, as is seen in human NCSLC patients with EGFr-L858R-driven lung tumors. Comparable studies with erlotinib, different tyrosine kinase inhibitors, and novel anti-cancer-causing agents ought to be accomplished in this and other informative GEM fashions to further investigate the tumor-preventative factors of those dealers.
5.4 Cancer vaccine
it is possible that sufferers with an excessive threat (own family history, environmental publicity, or heritable mutations) related to a specific cancer subtype would possibly gain from being
immunized at a younger age. Vaccine development relies upon the identification of antigens precise for a given cancer subtype or tumor-inducing biological agent. The heterogeneity and volatile genome in maximum cancers shows resistant mutant cells might gather speedy and triumph over the immunotherapy. However, vaccines against oncogenic strains of viruses have been validated to be very beneficial and often lean at the preclinical use of GEM models to expose the oncogenic potential of viral genes.
In 1993, studies of the use of transgenic mice emerged surely showing the oncogenic ability of the human papillomavirus (HPV) early genes E6 and E7 (Lambert et al., 1993; Arbeit et al., 1993). HPV DNA has also been discovered and hypothesized using a few to induce a subset of tongue and different oropharyngeal carcinomas (reviewed in Syrjänen, 2005). Transgenic mice expressing HPV early genes were used to illustrate the oncogenic potential of HPV in certain pores and skin cancers [HPV 8 (Schaper et al., 2005) and HPV 20 (Michel et al., 2006)].
Spurred by using these and many studies elucidating the connection between HPV and cervical most cancers, the FDA accredited the HPV vaccine Gardasil (Merck) for stopping the maximum commonplace sorts of human papillomavirus (HPV)-triggered cervical cancer (reviewed in McLemore, 2006). Feng et al., (2008) currently discovered a unique polyomavirus (similar to SV40) that included itself in the genome of 80% of the Merkel cellular carcinomas they tested. Although an extraordinary cancer, this is every other instance of a virus-related cancer that could be a target for most cancer-stopping vaccines and could be tested preclinical in GEM models
5.5 Legal Obstacle of preclinical testing in GEM models
In 1988 Harvard College filed the first of three exceptionally broad U.S. patents regarding the development and use of transgenic animals. Dupont (a sponsor of one of the Harvard investigators developing transgenic mice) became the extraordinary licensee of transgenic patents and merely sublicensed the patent rights (imposing huge charges and regulations) to enterprises and academia. This arrangement significantly confined the use of transgenic mice past primary discovery programs (reviewed in Sharpless & Depinho, 2006). Happily, For the greater medical network, the primary of those patents expired in 2005 and the second expired in February 2009; the 0.33 patent protecting trying out methods of the usage of transgenic oncomice is still under pressure through 2016. Hundreds of oncomice have been developed (normally in academia), but have not begun to be thoroughly studied and used on a big scale to test the growing quantity of feasible anti-cancer treatment plans. Once the very last restriction is lifted, researchers in industry, academia, and government alike will surely make bigger their use of transgenic models for preclinical drug research.
In preclinical anti-cancer drug development, Genetically Engineered Mouse models (gemstones) play a critical role in simulating human cancer biology. Gemstones are created with the aid of manipulating specific genes to either be overexpressed or knocked out, mainly due to the formation of tumors that resemble the ones discovered in human beings.
Commonplace methods consist of:
CRISPR-Cas9 technology introduces particular mutations that mimic human oncogenes or tumor suppressor gene loss.
Cre-Lox recombination for tissue-particular or time-specific gene alteration.
Tet-ON/OFF machine to manipulate gene expression dynamically using inducible elements inclusive of doxycycline.
Xenografts of human cancer cells into immunodeficient gemstones to observe tumor development and drug efficacy.
Gems are used to assess drug efficacy, recognize mechanisms of resistance, and expect drug toxicity through modeling one-of-a-kind cancer kinds, tiers, and genetic profiles.
Gems have confirmed achievement in diverse degrees of preclinical drug improvement: Identity of Drug Objectives: GEM fashions, inclusive of the ones harboring BRAF or EGFR mutations, have helped pick out powerful centered healing procedures (e.g., Vemurafenib for BRAF mutations in melanoma). Predicting Drug Efficacy: Tumor increase inhibition has been observed with novel inhibitors focused on oncogenic pathways (e.g., PI3K, HER2), offering sturdy predictive value for medical trials. Understanding Resistance Mechanisms: gemstones monitor how tumors evolve below drug strain, uncovering secondary mutations (e.g., inside the EGFR pathway) that lead to drug resistance. Toxicity Profiling: gems expressing humanized variations of drug targets permit for the prediction of potential off-target effects in everyday tissues, enhancing the protection profiles of candidate capsules.
Gemstones constitute a transformative tool in the preclinical segment of anti-cancer drug improvement. Their capacity to faithfully reflect cancer genetics and progression gives worthwhile insights into healing responses and resistance mechanisms. But, a few limitations include: Incomplete Human representation: gemstones, despite their utility, may not mimic the complexity of human cancers, particularly in regards to immune gadget interactions or tumor microenvironments. Inter-species Variability: Drug responses in gemstones won't always translate to humans due to variations in metabolism or genetic pathways. Cost and Time: The improvement and protection of gemstones are aid-in-depth, requiring sophisticated centers and know-how. However, gems keep playing a pivotal role in narrowing down drug candidates for clinical trials, providing greater particular, customized methods for cancer treatment.
To process the thousands of novel herbal products and artificial purported anti-most cancer compounds that arise each year, preclinical drug screens need to be as brief and complete as resources permit. This can be accomplished by way of in vitro displays the use of cultured cellular lines that have oncogenic gene expression profiles similar to the ones visible in human patients. Compounds that display activity can then be moved to xenograft studies, which check toxicity and tumor regression ability. even though we've reviewed the energy of GEM models in verifying drug efficacy and pathway mechanism analysis, GEM fashions as they exist nowadays have fundamental flaws that prevent their use as high-throughput drug-screening tools. Gemstones require numerous months to develop tumors, and the penetrance is regularly far less than a hundred%. Tumors rise and grow at distinct costs in every man or woman mouse in a given observation. Besides melanoma, breast cancer, and some prostate cancer models where palpable tumors broaden, figuring out which animals have developed tumors at any given moment turns into a nearly insurmountable assignment. Many published studies use MRI pics to comply with tumor progression. Considering imaging the thorax of one mouse in an MRI can soak up to one hour, this method is incompatible with huge, excessive throughput studies. Small animal in vivo imaging (SAIVI) the usage of fluorescent or bioluminescent tagged sellers concentrated on tumors is a promising opportunity, however, the technology continues to be in its infancy, lacks suitably inducible GEM models for most varieties of cancers, and often require buy of luxurious, photosensitive probes and detection systems. Computed Tomography (CT) Imaging with medical-length instruments cannot be used due to the fact the dose of radiation used at some stage in imaging may additionally have healing effects on the tumors, especially when GEM models will require multiple imaging sessions over their lifetime to first pick out a tumor and then to monitor therapeutic drug consequences. But as smaller, less powerful, rodent-particular CT imagers become to be had by researchers, this can prove to be a beneficial method to degree tumor length in deep tissue. To make use of cutting-edge GEM models in preclinical drug trials, conventional tumor staging techniques have to be revised for every mouse to be treated as an affected person in a medical trial. Healing agents have to now not be administered en mass to all individuals in a treatment institution at a given time factor, but tailor-made to each mouse reflecting the date the tumor was recognized by using something modality the researcher has evolved. This can create a logistical nightmare, particularly if big combination treatment trials trying out multiple cars, routes of management, dosing schedules, and a couple of dose concentrations are attempted. Day zero has to be marked at the time an animal’s measured tumor length or general “tumor burden” (if the GEM generates a couple of foci concurrently) crosses a pre-determined threshold amount. Therefore, we cannot envision GEM fashions changing in vitro and xenograft models for the high throughput efficacy displays of novel dealers. But, we do trust GEM fashions can be of amazing value in the course of 3 steps within the preclinical process (discern 1). First, if GEM tumors are harvested and fragments efficaciously implanted subcutaneously (SC) or orthotopically into syngeneic, immune-ready mice, sufficient animals can be accrued to perform conventional xenograft-like monitors with genetically suitable murine tumors (allografts). Varticovski et al., (2007) offer an instance of this method, harvesting MMTV-PyMT breast tumors from some mice and passing them as fragments or cells suspensions into numerous host animals for subsequent drug studies. DNA microarrays have been used to affirm the handiest slight adjustments in gene expression after serial passages from the unique tumor. 2nd passage tumors exhibited similar sensitivity to paclitaxel and cyclophosphamide in comparison to the original tumor material. Once an agent suggests activity in vitro and tumor transplants, GEM models can be used to explore the drug mechanism and further optimize in vivo dosing before an agent head into an awful lot of extra expensive and onerous scientific trials (discern 1). Those optimization studies want now not to have excessive throughput and can take a year or more to finish. As a result, fewer anti-cancer-causing agents will efficaciously bypass those additional preclinical reviews than do presently develop to clinical trials. Currently, the handiest ~5% of novel anti-carcinogens entering scientific trials gain FDA approval (Kola & Landis, 2004); the maximum value and attrition takes place during section II and section III clinical trials. Consequently, any more time GEM-based total drug screening would upload to the preclinical drug development pipeline should be financially worthwhile to the drug sponsors and ethically useful for the volunteers participating in clinical trials if more efficacious cures are determined. GEM models might also in the long run have a fourth function in preclinical drug testing if researchers can effectively “humanize” them to be used in toxicology and pharmacology research. The cytochrome P450 (CYP) family of enzymes is expressed ordinarily in the liver and is worried inside the metabolism of a numerous range of healing compounds, pollutants, carcinogens, hormones, and xenobiotic dealers we may also come across. The seven most important CYP gene clusters found in humans are gifted and increased in mice (57 putative human CYP genes versus 102 putative useful genes in mice) (Nelson et al., 2004). But studies have shown that many character CYPs are differentially expressed or differentially active among the mouse and humans (Bogaards et al., 2000). This explains why drug metabolism inside the mouse does now not continually mirror and is expecting drug metabolism and toxicity within the human medical trials. Numerous GEM fashions have been generated in which the endogenous mouse CYPs or different xenobiotic-related metabolism genes are deleted and changed with their human orthologues, or without a doubt over explicitly the human orthologue in addition to the mouse CYP (reviewed in Cheung & Gonzalez, 2008). The expression of one or two human genes certainly is now not sufficient to declare a capacity GEM model as functionally “humanized” and geared up for excessive throughput toxicity and pharmacology research. But as transgenic technology evolves and these mice specifically several human orthologues, will indeed be able to higher predict human drug hobby and toxicity, and be of crucial importance in preclinical healing drug improvement.
The authors would like to acknowledge the contributions and support of individuals and organizations who aided in the completion of this research project. Special thanks to My Mentor Naweed Imam Syed Prof. Department of Cell Biology at the University of Calgary and Dr. Sadaf Ahmed from the Psychophysiology Lab at the University of Karachi for their invaluable input and support throughout the research.
Declaration of Interest:
The authors declare no conflicts of interest or financial disclosures related to this research.
Financial Support and Sponsorship:
No funding was received for the preparation of this manuscript.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
